Step 1: Locally Optimise Reactants and Products¶
Read the theory first
Before starting this step, we recommend reading the theory for this step to understand why we are performing this step.
- Make sure you have also read the theory on the potential energy surface.
Tip
Click the Download Templates.zip button to download the template folder for this step.
- If you would like to follow this guide using data already calculated using ORCA, click
Download Examples.zip.
First, we need to locally optimise the reactants and products. To do this:
- Make a
Reactantsand aProductsfolder - Create a new folder for each reactant and product in your mechanistic step.
- Add the
.inpfile to each folder for performing local optimisations. Make sure you include the following into your.inpfiles for all your reactants and products:
Here, we perform a geometry optimisation to optimise the system. The tags here indicate you want to do the following:
OPT: Indicates you want ORCA to perform a local optimisation.FREQ: Indicates you want ORCA to calculate the vibrational frequency for your molecule.- This is used to verify that your optimised structure is indeed a local minimum.
- This will also give you the Gibbs free energy for your molecule that you (may) want to report as your energy.
TightOPT: Tells ORCA to tighten the convergence criteria for each geometric step. See the ORCA 6.1.0 Manual, page 618 for more info.TightSCF: Tells ORCA to tighten the convergence criteria for each electronic step.defgrid2: Indicates how fine we want the integration grid to be (this is the default).
NOTE 1
I have set the electronic optimisation steps to be tight (TightSCF). This is just to make sure the electronic steps are well converged, but it may be a bit extreme.
- If you have problems, you can try using the normal convergence criteria for the electronic steps (
NormalSCF)
NOTE 2
Click here for more information about other electronic convergence and integration grid settings (besides TightSCF and defgrid2)
An example of the complete orca.inp file for a local optimisation ORCA job is given below (also located here):
!B3LYP DEF2-TZVP D3BJ
!OPT FREQ TightOPT TightSCF defgrid2
%SCF
MaxIter 2000 # Here setting MaxIter to a very high number. Intended for systems that require sometimes 1000 iterations before converging (very rare).
DIISMaxEq 5 # Default value is 5. A value of 15-40 necessary for difficult systems.
directresetfreq 15 # Default value is 15. A value of 1 (very expensive) is sometimes required. A value between 1 and 15 may be more cost-effective.
END
%CPCM EPSILON 6.02 REFRAC 1.3723 END
%PAL NPROCS 32 END
%maxcore 2000 # This indicates you want ORCA to use only 2GB per core maximum, so ORCA will use only 2GB*32=64GB of memory in total.
* xyzfile 1 1 product.xyz
NOTE 3
Make sure you include a newline or two at the end of your orca.inp file, otherwise ORCA will get confused and not run.
The last line of the ORCA inp file contains the following:
- The input type for inputting the coordinates of your system.
- The overall charge of your system.
- The overall multiplicity of your system.
- If you use the
xyzfiletype, the xyz file of your system.
Here, xyzfile allows you to import an xyz file into ORCA. You can add the xyz data directly in the .inp file, but I find having a separate xyz file is better because this allows you to look at the xyz file in a GUI like in atomic simulation environment (ASE).
- Include the
xyzfiles of your reactant and product molecules in theReactantandProductfolders, respectively. - If your reactants or products contain more than one molecule/chemical system, split them up and optimise them individually in their own individual folders.
What is the multiplicity of my system?
The multiplicity indicates what the total spin of the electrons in your system (reactants or products) is. This is based on how many electrons are in your system, where the multiplicity is based on the \(M = 2S + 1\) rule (and where \(S\) is the total spin of your electrons):
- If you have an even number of electrons in your system, the multiplicity will be an odd number. The lowest value multiplicity is 1.
- If you have an odd number of electrons in your system, the multiplicity will be an even number. The lowest value multiplicity is 2 (It can not be 0).
If you want to easily figure out if you should have an even or odd multiplicity, you can use:
where
filepathis the path to thexyzfile of your system, and--chargeis the overall charge of your system (if--chargeis not given, the default given is 0).
See more about viewORCA multi by clicking here for the multi entry in the viewORCA manual.
Note: Your multiplicity may be dependent on if you have transition metals in your system. For example, 3d transition metals can have valence electrons in degenerate energy levels.
Examples: Based on crystal field theory, octahedral transition metal complexes have 3 lower energy t2g orbitals, and 2 higher energy eg orbitals. Thinking about this orbital scheme, consider the following examples:
-
Example 1: Octahedral Cu(I) has 10 valence electrons in the 3d orbitals. 6 of these electrons live in the t2g, while the other 4 electrons live in the eg orbitals. Therefore, the total electron spin is \(0\), giving a multiplicity of \(M = 2(0) + 1 = 1\).
-
Example 2: Octahedral Cu(II) complexes have 9 valence electrons. 6 of these electrons live in the t2g, while the other 3 electrons live in the eg orbitals. Therefore, the total electron spin is \(1/2\), giving a multiplicity of \(M = 2(1/2) + 1 = 2\).
-
Example 3: Octahedral Fe(III) complexes have 5 valence electrons. In the high spin case, 3 of these electrons live individually in each of the 3 t2g, while the other 2 electrons live in the 2 eg orbitals. Therefore, the total electron spin is \(5/2\), giving a multiplicity of \(M = 2(5/2) + 1 = 6\).
Submit the job to slurm (if you use slurm) using the submit.sl file:
#!/bin/bash -e
#SBATCH --job-name=A3_Step1_Products
#SBATCH --ntasks=32
#SBATCH --mem-per-cpu=2500MB
#SBATCH --partition=genoa,milan
#SBATCH --time=3-00:00 # Walltime
#SBATCH --output=slurm-%j.out # %x and %j are replaced by job name and ID
#SBATCH --error=slurm-%j.err
#SBATCH --nodes=1 # OpenMPI can have problems with ORCA over multiple nodes sometimes, depending on your system.
# Load ORCA
module load ORCA/6.1.0-f.0-OpenMPI-4.1.5
# ORCA under MPI requires that it be called via its full absolute path
orca_exe=$(which orca)
# Don't use "srun" as ORCA does that itself when launching its MPI process.
${orca_exe} orca.inp > output.out
NOTE 4
While ORCA has been told to use maxcore = 2000 (MB) * 32 = 64 GB in the .inp file, we have told slurm to reserve 2500MB * 32 = 80 GB of memory. It is a good idea to give your ORCA job a bit of extra memory in slurm just in case ORCA accidentally goes over its allocated RAM. Here, I have arbitrarily given this job 500 MB more RAM per CPU just in case.
Outputs from ORCA for Example¶
While ORCA is running, it will output several files, including an output.out file, an orca.xyz file, and an orca_trj.xyz file.
output.out: This file contains the details about how ORCA ran. This includes the vibrational frequency data to check if the locally optimised structure is in fact a local minimum.orca.xyz: This is the locally optimised molecule.orca_trj.xyz: This file shows how ORCA locally optimised the molecule. TypeviewORCA optinto the terminal to view how the molecule was optimised, including an energy profile.
Once ORCA has finished, you should do the following checks:
Check 1: Look at your molecule and the energy profile and make sure it looks ok¶
The first thing to do is to look at your molecule and check if it looks sensible with your chemical intuition. You can do this by opening up the orca.xyz in your favourite GUI. I like to use atomic simulation environment (ASE). To look at the molecule and its energy profile:
- Open a new terminal
cdinto the optimisation folder- Type
viewORCA optinto the terminal
# cd into your optimisation folder
cd ORCA_Mechanism_Procedure/Example/Step1_Geo_Opt/Products
# View the optimisation
viewORCA opt
NOTE 1
If viewORCA opt does not work, type ase gui orca_trj.xyz into the terminal instead of viewORCA opt.
NOTE 2
viewORCA opt will also create a xyz file called OPT_images.xyz that you can copy and view on your own computer.
- If you just want to create the
OPT_images.xyzfile and not view the optimisation, type into the terminalviewORCA opt --view False(which will create theOPT_images.xyzfile).
Here, you want to check that the molecule looks ok from your chemical and physical intuition. Here is an example of what the optimised molecule looks like (the orca.xyz file here). If we look at the initial molecule geometry (by typing viewORCA opt or ase gui product.xyz into the terminal), we can see how the molecule has changed after being geometrically optimised:

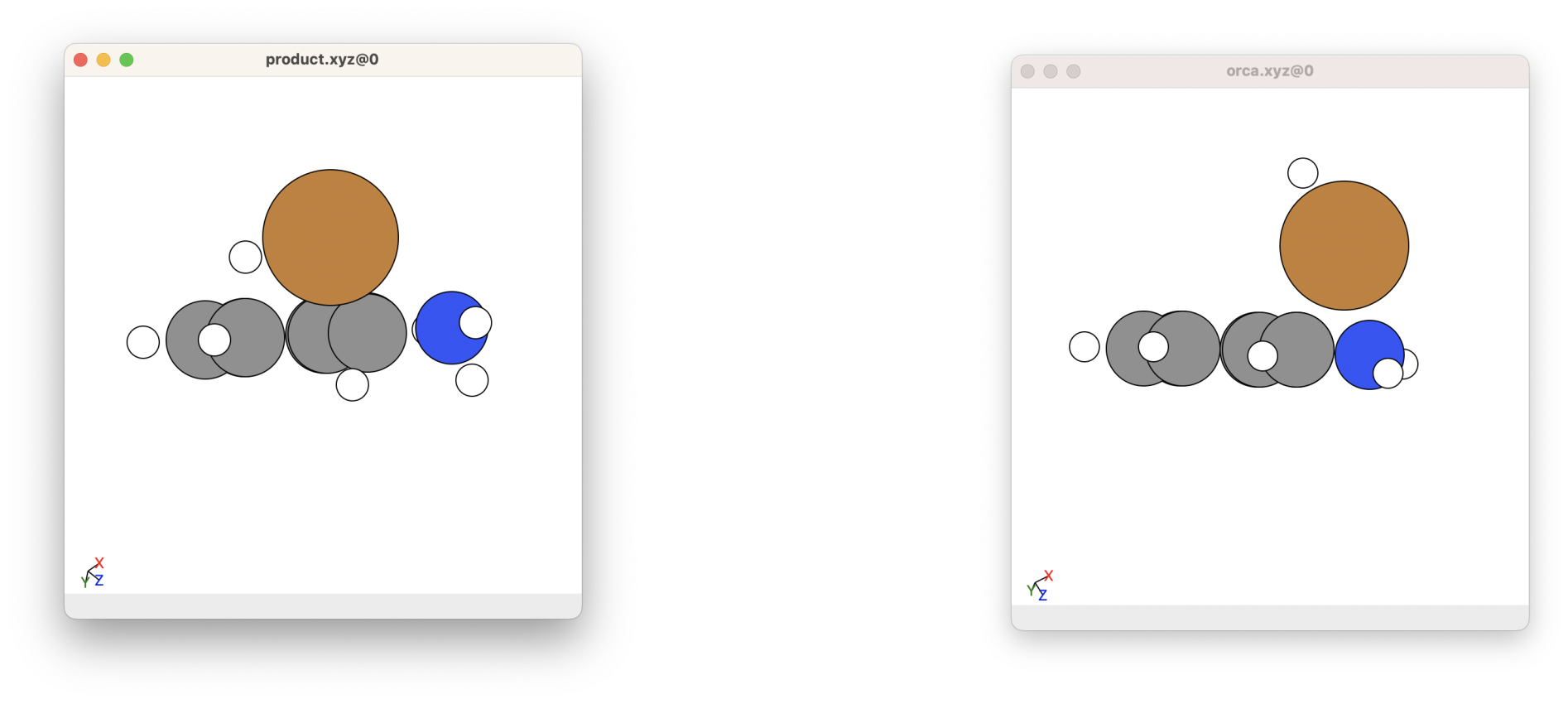
viewORCA opt will also show you the energy profile for this optimisation.
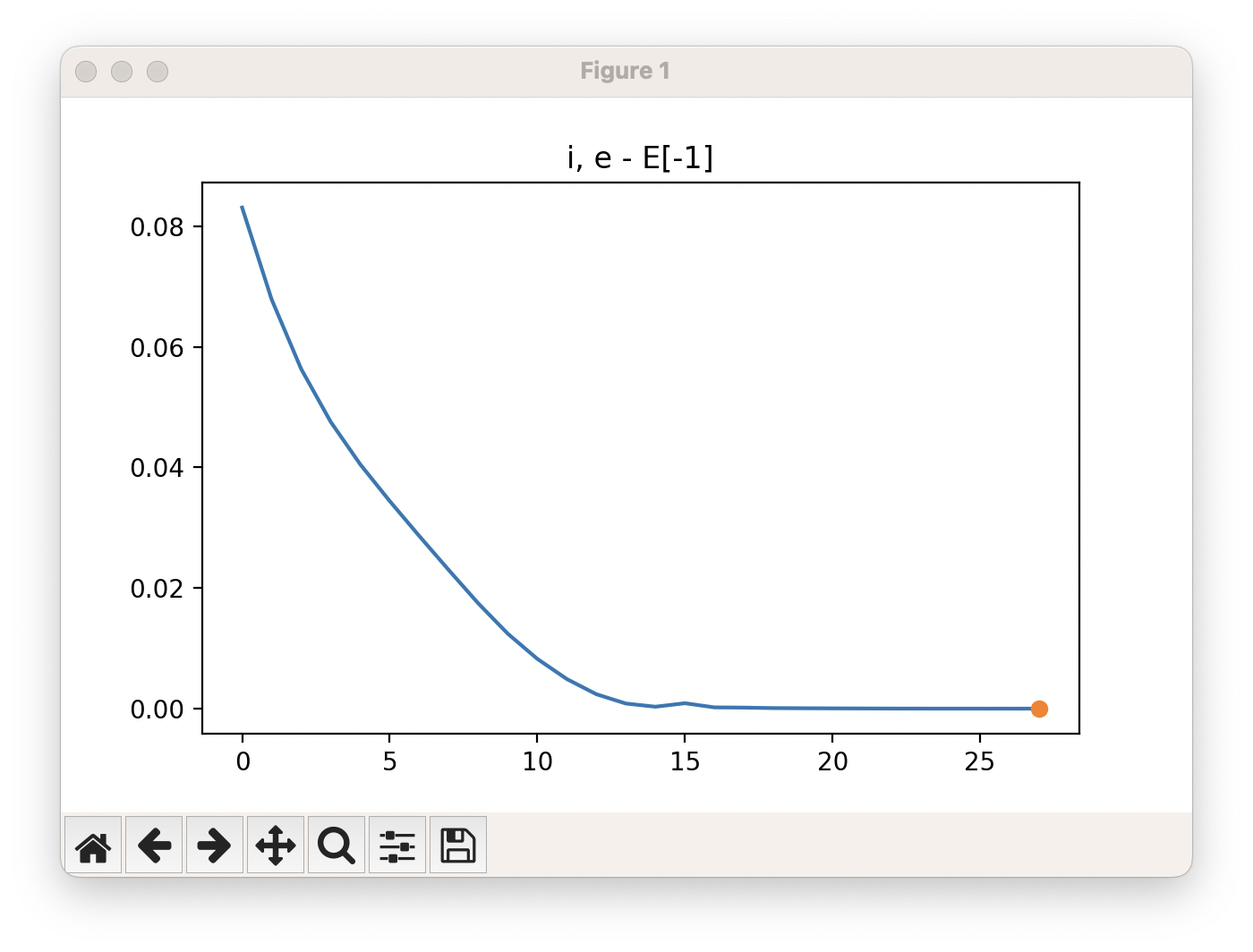
Check 2: Did the geometry optimisation converge successfully¶
You want to look in the output.out file for a table with the title Geometry convergence. There will be many of these tables, as one is given for each geometric step performed.
- You want to look at the last
Geometry convergencetable in the output file as this will display the information for the latest geometrically optimised step. An example for theProductsis given below:
.--------------------.
----------------------|Geometry convergence|-------------------------
Item value Tolerance Converged
---------------------------------------------------------------------
Energy change -0.0000121554 0.0000010000 NO
RMS gradient 0.0000126838 0.0000300000 YES
MAX gradient 0.0000468077 0.0001000000 YES
RMS step 0.0001259501 0.0006000000 YES
MAX step 0.0005455187 0.0010000000 YES
-------------------------------------------------------------------------
........................................................
Max(Bonds) 0.0000 Max(Angles) 0.01
Max(Dihed) 0.03 Max(Improp) 0.00
---------------------------------------------------------------------
Everything but the energy has converged. However, the energy
appears to be close enough to convergence to make sure that the
final evaluation at the new geometry represents the equilibrium energy.
Convergence will therefore be signaled now
***********************HURRAY********************
*** THE OPTIMIZATION HAS CONVERGED ***
*************************************************
In this example, you can see that the majority of the items of interest have converged, and ORCA is happy that the convergence criteria have been met. ORCA also tells you this by giving you a HURRAY message as well as a THE OPTIMIZATION HAS CONVERGED message (as you can see above).
Check 3: Check that the molecule has no negative vibrational frequencies¶
After performing a local optimisation, it is important that you look at the vibrational frequencies that are calculated. These are the frequencies that you could see in an IR or Raman spectrum. You want to look through your ORCA output.out file for a heading called VIBRATIONAL FREQUENCIES. We want to make sure that all the frequencies are non-negative. This means we are in a local energy well (see the Theory page for why this works).
- If one or more frequencies are negative, this means we are not in a local minimum. In this case, you need to tighten the optimisation, or need to look at your molecule and see if any part of it structurally does not make sense with your chemical intuition.
In the example below (for Products), you can see that there are no negative frequencies from the FREQ calculation, therefore we are in a local energy well:
-----------------------
VIBRATIONAL FREQUENCIES
-----------------------
Scaling factor for frequencies = 1.000000000 (already applied!)
0: 0.00 cm**-1
1: 0.00 cm**-1
2: 0.00 cm**-1
3: 0.00 cm**-1
4: 0.00 cm**-1
5: 0.00 cm**-1
6: 44.93 cm**-1
7: 69.32 cm**-1
8: 145.14 cm**-1
9: 225.50 cm**-1
10: 280.47 cm**-1
11: 304.99 cm**-1
12: 391.81 cm**-1
13: 402.73 cm**-1
14: 415.60 cm**-1
15: 472.47 cm**-1
16: 496.43 cm**-1
17: 584.01 cm**-1
18: 628.47 cm**-1
19: 679.74 cm**-1
20: 722.32 cm**-1
21: 784.15 cm**-1
22: 793.27 cm**-1
23: 809.82 cm**-1
24: 845.31 cm**-1
25: 953.98 cm**-1
26: 980.92 cm**-1
27: 997.22 cm**-1
28: 1018.49 cm**-1
29: 1030.84 cm**-1
30: 1046.46 cm**-1
31: 1103.59 cm**-1
32: 1137.99 cm**-1
33: 1187.92 cm**-1
34: 1210.32 cm**-1
35: 1238.50 cm**-1
36: 1312.26 cm**-1
37: 1358.75 cm**-1
38: 1377.48 cm**-1
39: 1481.29 cm**-1
40: 1515.76 cm**-1
41: 1542.98 cm**-1
42: 1608.70 cm**-1
43: 1632.82 cm**-1
44: 1646.95 cm**-1
45: 1845.23 cm**-1
46: 3184.93 cm**-1
47: 3187.86 cm**-1
48: 3191.70 cm**-1
49: 3193.64 cm**-1
50: 3204.14 cm**-1
51: 3209.64 cm**-1
52: 3487.75 cm**-1
53: 3585.96 cm**-1
Troubleshooting Geometric Optimisation (Opt) Calculations¶
Here are some troubleshooting tips for performing this optimisation step.
Note
Also read more about electronic convergence issues that are commonly encountered during geometric optimisation jobs from the ORCA Input Library by clicking here.
- The ORCA Input Library is generally a great source of information about performing calculations in ORCA.
Info
If you have any issues about this step, raise a New issue at the Issues section by clicking here.
ORCA would not converge but reached the maximum number of optimization cycles.¶
A common problem that can arise is the message from ORCA saying that it could not converge because it had reached the maximum number of optimisation cycles.
----------------------------------------------------------------------------
ERROR !!!
The optimization did not converge but reached the maximum
number of optimization cycles.
As a subsequent Frequencies calculation has been requested
ORCA will abort at this point of the run.
Please restart the calculation with the lowest energy geometry and/or
a larger maxiter for the geometry optimization.
----------------------------------------------------------------------------
This indicates that the electronic step could not converge for the last geometric step. This means that ORCA could not find a way to optimise the molecular orbitals for this geometric step. This is a very common issue. In this case, you can replace your initial structure with the last optimised structure given by ORCA. Do this by changing the name of the input file in the orca.inp file from reactant.xyz to orca.xyz
However, before proceeding you should check the initial structure you gave to ORCA.
- The
converge but reached the maximum number of optimization cycles.problem can arise because the initial structure you gave to ORCA was not a good starting point. - ORCA requires that you apply your chemical intuition to your system before you run it. Otherwise it can fail.
For example, consider that we want to optimise the following chemical system, where Cu+ coordinates to the nitrogen in benzylamine:
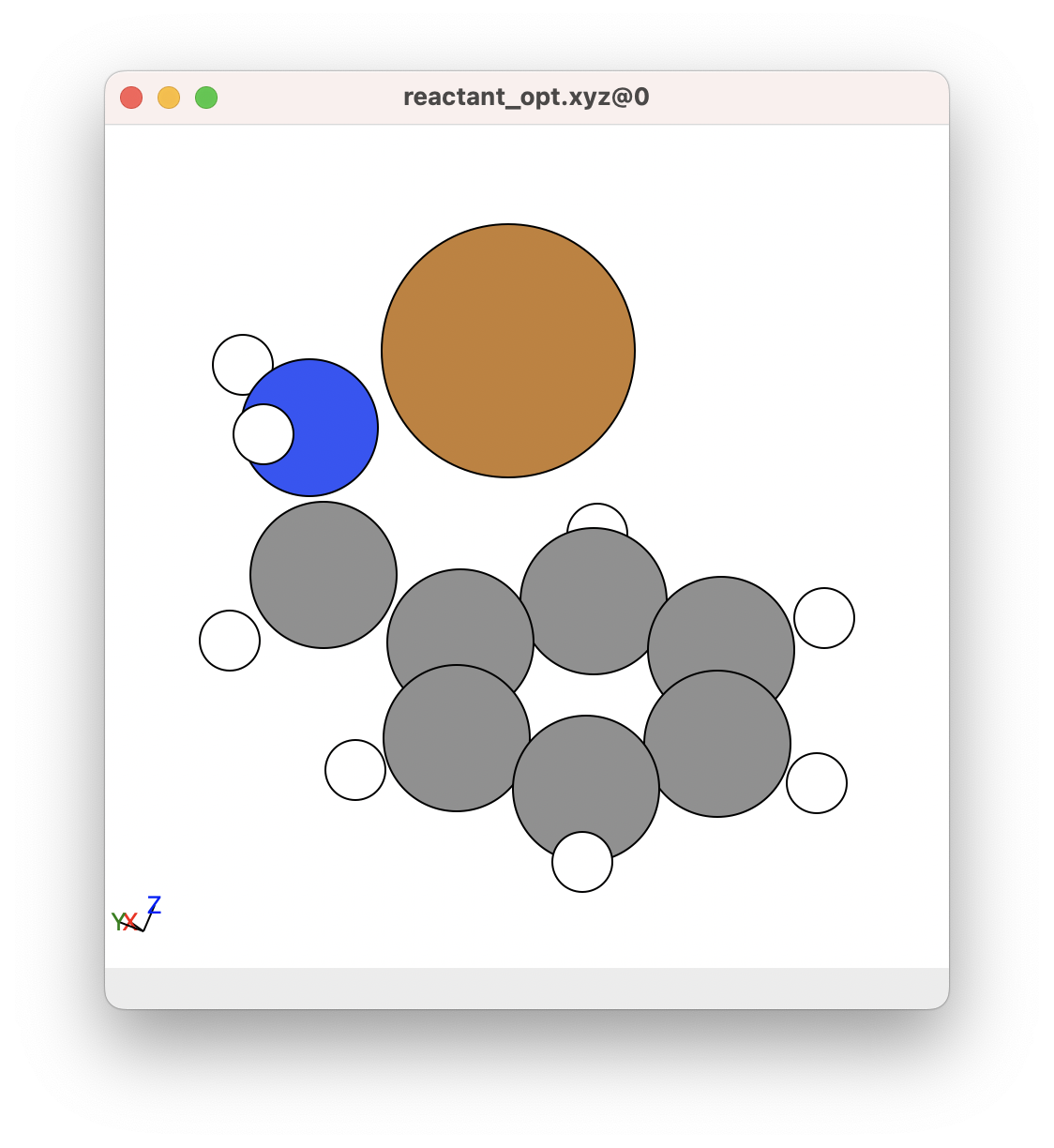
If we wanted to obtain this chemical system, we would want to make sure that most of the atoms are roughly in a good bonding position. This is a good example of a good structure to begin with:
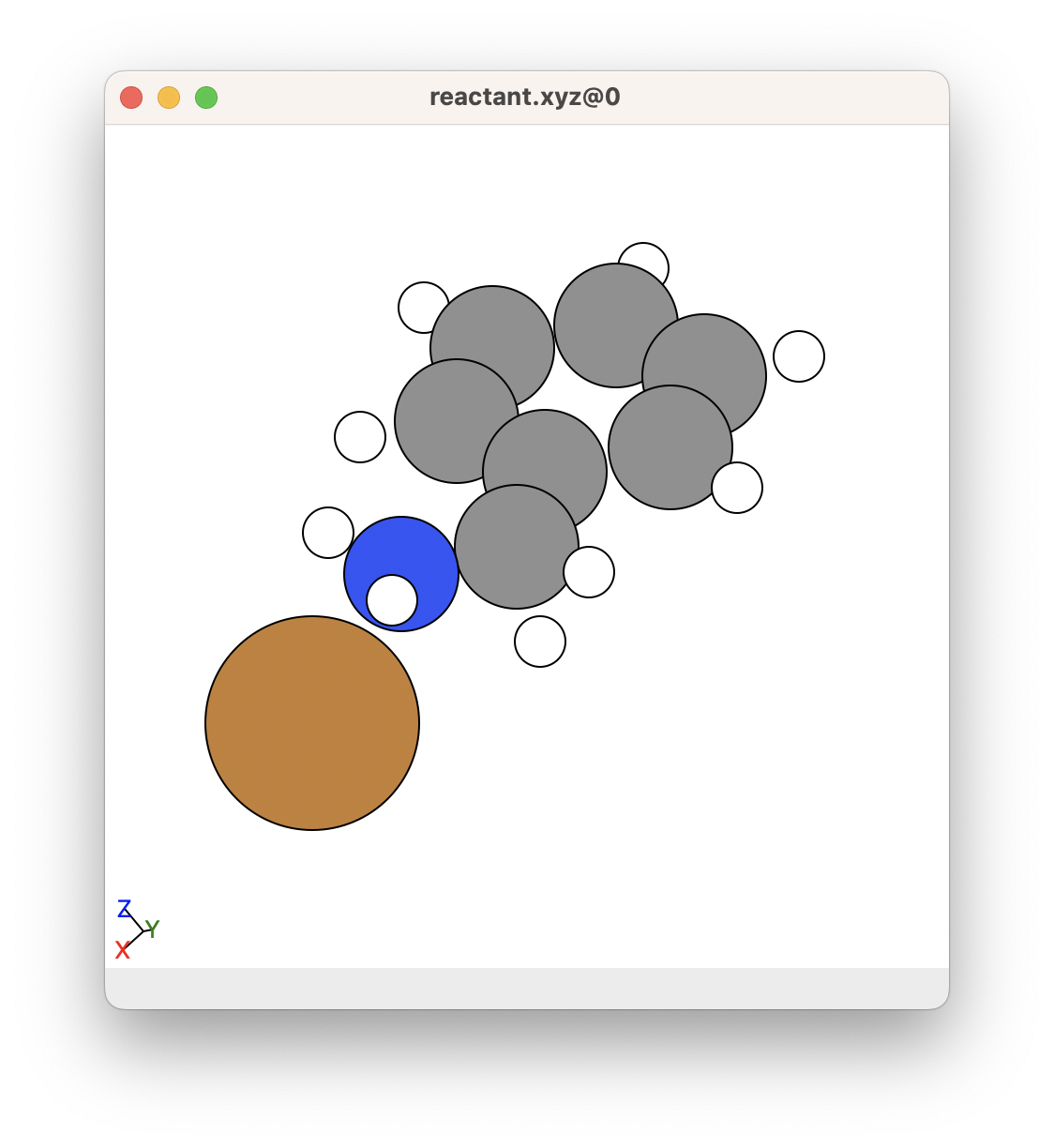
Below is an example of a structure that might have convergence issues because the Cu+ atom is very far away from the nitrogen. Generally, it is best not to combine separate molecules/compounds in an ORCA calculation unless they are well separated and are not meant to come together for geometry optimisations. This does not apply to SCAN calculations, where you might want to separate two components from each other (For example, separate Cu+ from benzylamine). We will talk about SCAN calculations in the next section.

In this case, it would be best to try using a better initial structure where the Cu+ atom is coordinated to the benzylamine molecule.