Step 4B: Check the Forwards and Backwards Molecules Converge using the TightOPT or NormalOPT tags¶
Tip
Click the Download Templates.zip button to download the template folder for this step.
- If you would like to follow this guide using data already calculated using ORCA, click
Download Examples.zip.
In Step 4A we found the local wells that mechanistically connect to each other through our transition state using the TolMAXG and TolRMSG tags equivalent to the LooseOPT tag. However, it is best to make sure that our forwards and backwards steps converge using the TightOPT or NormalOPT tags, and perform a vibrational frequency calculation to confirm our molecules have fallen into a local energy well.
- We will then compare this with our reactant and product molecules from Step 1 to make sure that this mechanistic step is in fact the mechanistic step we are wanting to get the transition state for.
To do this, we will repeat Step 1 for our forwards and backwards molecules. To do this, make a Forwards folder and a Backwards folder, and add to each folder the .inp file for performing local optimisations. Make sure you include the following into your .inp files for both the forwards and backwards input files:
Here, we perform a geometry optimisation to optimise the system. The tags here indicate you want to do the following:
OPT: Indicates you want ORCA to perform a local optimisation.FREQ: Indicates you want ORCA to calculate the vibrational frequency for your molecule. This is used to verify that your optimised structure is indeed a local minimum. This will also give you the Gibbs free energy for your molecule that you (may) want to report as your energy.TightOPT: Tells ORCA to tighten the convergence criteria for each geometric step. See ORCA 6.1.0 Manual, page 618 for more info.TightSCF: Tells ORCA to tighten the convergence criteria for each electronic step.defgrid2: Indicates how fine we want the integration grid to be (this is the default).
Also include the xyz files of your forwards (orca_IRC_F.xyz) and backwards (orca_IRC_B.xyz) molecules from Step 4A in your Forwards and Backwards folders, respectively.
An example of the complete orca.inp file for a local optimisation ORCA job is given below (also located here):
!B3LYP DEF2-TZVP D3BJ
!OPT FREQ TightOPT TightSCF defgrid2
%SCF
MaxIter 2000 # Here setting MaxIter to a very high number. Intended for systems that require sometimes 1000 iterations before converging (very rare).
DIISMaxEq 5 # Default value is 5. A value of 15-40 necessary for difficult systems.
directresetfreq 15 # Default value is 15. A value of 1 (very expensive) is sometimes required. A value between 1 and 15 may be more cost-effective.
END
%CPCM EPSILON 6.02 REFRAC 1.3723 END
%PAL NPROCS 32 END
%maxcore 2000 # This indicates you want ORCA to use only 2GB per core maximum, so ORCA will use only 2GB*32=64GB of memory in total.
* xyzfile 1 1 orca_IRC_F.xyz
Outputs from ORCA¶
When ORCA is running, it will output several files, including an output.out file, an orca.xyz file, and an orca_trj.xyz file.
output.out: This file contains the details about how ORCA ran. This includes the vibrational frequency data to check if the locally optimised structure is in fact a local minimum.orca.xyz: This is the locally optimised molecule. This is the file that is calledreactant_opt.xyzfrom here on out.orca_trj.xyz: This file shows how ORCA locally optimised the molecule. TypeviewORCA optinto the terminal to view how the molecule was optimised, including an energy profile.
Once ORCA has finished, you should do the following checks. These are the same checks as we performed in Step 1 (shown is an example for the Forwards step; also repeat this process on the Backwards step):
Check 1: Look at your molecule and the energy profile and make sure it looks ok¶
The first thing to do is to look at your molecule and check if it looks sensible with your chemical intuition. You can do this by opening up the orca.xyz in your favourite GUI. I like to use atomic simulation environment (ASE). To look at the molecule and its energy profile:
- Open a new terminal
cdinto the optimisation folder- Type
viewORCA optinto the terminal
# cd into your optimisation folder
cd ORCA_Mechanism_Procedure/Examples/Step4_Validate_TS/Step4B_Validate_Forwards_and_Backwards/Forwards
# View the optimisation
viewORCA opt
NOTE 1
If viewORCA opt does not work, type ase gui orca_trj.xyz into the terminal instead of viewORCA opt.
NOTE 2
viewORCA opt will also create a xyz file called OPT_images.xyz that you can copy and view on your own computer.
- If you just want to create the
OPT_images.xyzfile, type into the terminalviewORCA opt --view False(which will create theOPT_images.xyzfile without showing thease guiwindow).
Here, you want to check that this molecule looks like either the product or reactant. In this example, we can see that this molecule is equivalent to the product, where the Cu atom has inserted itself into the C-H bond:


viewORCA opt will also show you the energy profile for this optimisation.
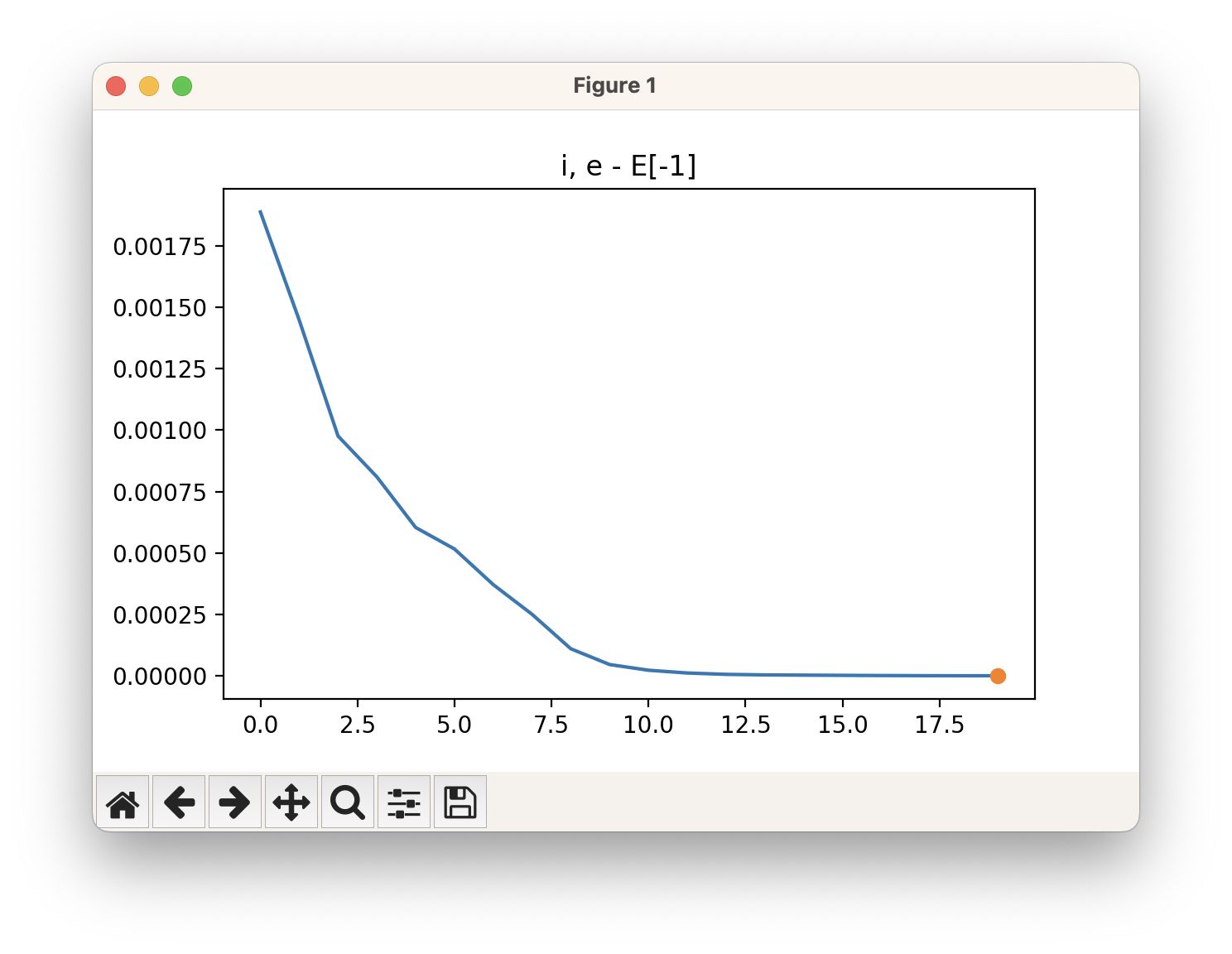
Check 2: Did the geometry optimisation converge successfully¶
You want to look in the ORCA output.out file for a table with the title Geometry convergence. There will be many of these tables, as one is given for each geometric step performed. You want to look at the last Geometry convergence table as this will give the details for the latest geometrically optimised step. An example for the Forwards is given below:
.--------------------.
----------------------|Geometry convergence|-------------------------
Item value Tolerance Converged
---------------------------------------------------------------------
Energy change 0.0000004576 0.0000010000 YES
RMS gradient 0.0000125618 0.0000300000 YES
MAX gradient 0.0000744173 0.0001000000 YES
RMS step 0.0002245851 0.0006000000 YES
MAX step 0.0007713970 0.0010000000 YES
-------------------------------------------------------------------------
........................................................
Max(Bonds) 0.0003 Max(Angles) 0.03
Max(Dihed) 0.04 Max(Improp) 0.00
---------------------------------------------------------------------
***********************HURRAY********************
*** THE OPTIMIZATION HAS CONVERGED ***
*************************************************
In this example, you can see that the majority of the items of interest have converged, and ORCA is happy that the convergence criteria have been met. ORCA also tells you this by giving you a HURRAY message as well as a THE OPTIMIZATION HAS CONVERGED message (as you can see above).
Check 3: Check that the molecule has no negative vibrational frequencies¶
After performing a local optimisation, it is important that you look at the vibrational frequencies that were calculated in the ORCA output file. These are the frequencies that you could see in an IR or Raman spectrum. You want to look through your .out file for a heading called VIBRATIONAL FREQUENCIES. We want to make sure that all the frequencies are non-negative. This means we are in a local energy well (see the Theory page for why this works). If one or more frequencies are negative, this means we are not in a local minimum. In this case, you need to tighten the optimisation, or need to look at your molecule and see if any part of it structurally does not make sense with your chemical intuition.
In the example below (for Forwards), you can see that there are no negative frequencies from the FREQ calculation, therefore we are in a local energy well:
-----------------------
VIBRATIONAL FREQUENCIES
-----------------------
Scaling factor for frequencies = 1.000000000 (already applied!)
0: 0.00 cm**-1
1: 0.00 cm**-1
2: 0.00 cm**-1
3: 0.00 cm**-1
4: 0.00 cm**-1
5: 0.00 cm**-1
6: 46.05 cm**-1
7: 70.06 cm**-1
8: 146.05 cm**-1
9: 225.14 cm**-1
10: 281.70 cm**-1
11: 305.43 cm**-1
12: 391.89 cm**-1
13: 402.99 cm**-1
14: 415.38 cm**-1
15: 472.44 cm**-1
16: 496.67 cm**-1
17: 584.22 cm**-1
18: 628.51 cm**-1
19: 679.99 cm**-1
20: 721.45 cm**-1
21: 784.71 cm**-1
22: 794.19 cm**-1
23: 809.89 cm**-1
24: 845.55 cm**-1
25: 954.05 cm**-1
26: 980.23 cm**-1
27: 997.24 cm**-1
28: 1018.49 cm**-1
29: 1030.82 cm**-1
30: 1046.51 cm**-1
31: 1103.69 cm**-1
32: 1138.20 cm**-1
33: 1187.91 cm**-1
34: 1210.53 cm**-1
35: 1238.59 cm**-1
36: 1312.08 cm**-1
37: 1358.82 cm**-1
38: 1377.79 cm**-1
39: 1481.33 cm**-1
40: 1515.59 cm**-1
41: 1542.97 cm**-1
42: 1608.75 cm**-1
43: 1632.94 cm**-1
44: 1646.78 cm**-1
45: 1845.36 cm**-1
46: 3185.00 cm**-1
47: 3187.95 cm**-1
48: 3191.67 cm**-1
49: 3193.66 cm**-1
50: 3204.18 cm**-1
51: 3209.66 cm**-1
52: 3488.03 cm**-1
53: 3586.23 cm**-1
Example of an issue with Step 4 in this case, but all is well.¶
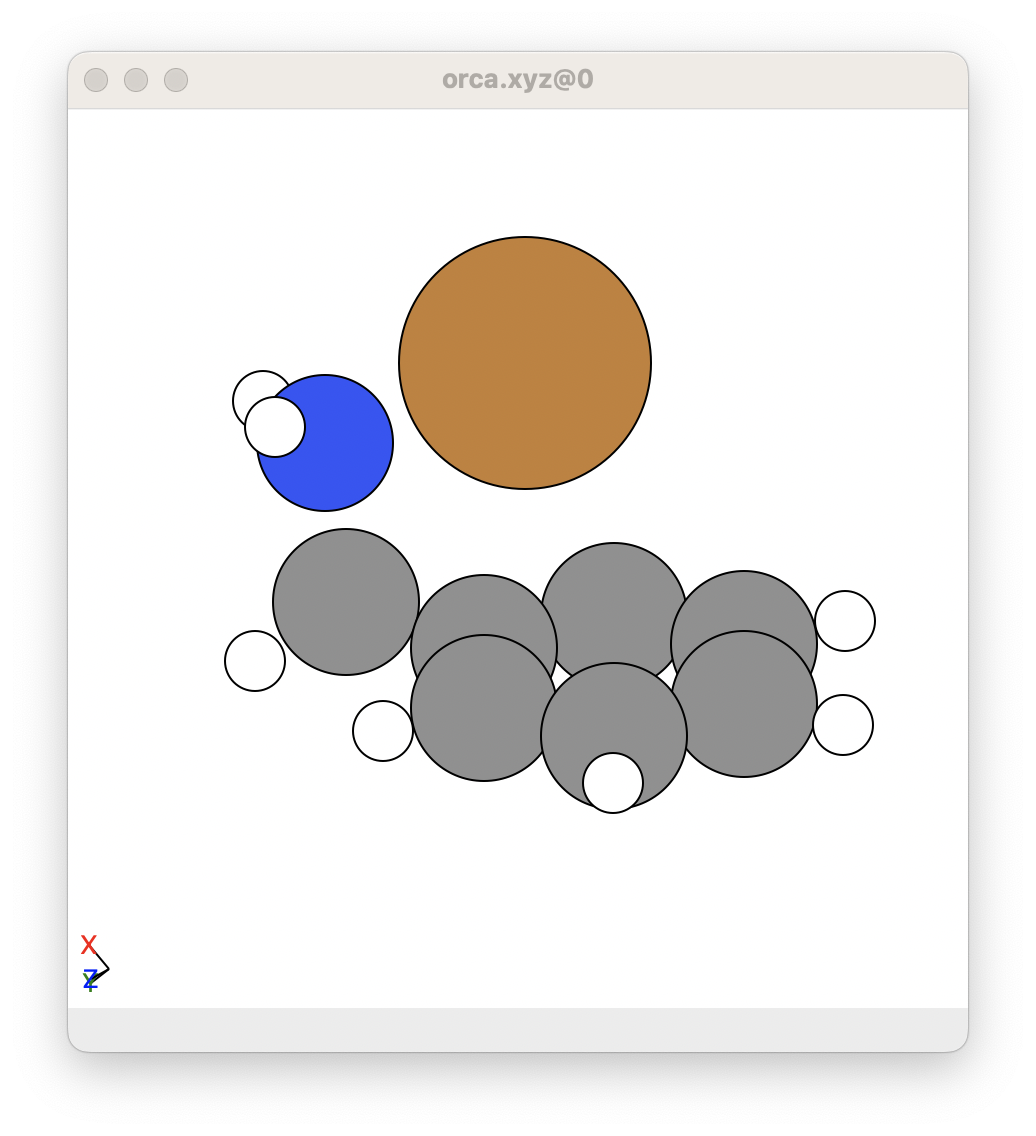
In this example, the optimised reactant from Step 1 looks like this:
However, we get the following for the backwards structure from Step 4A.
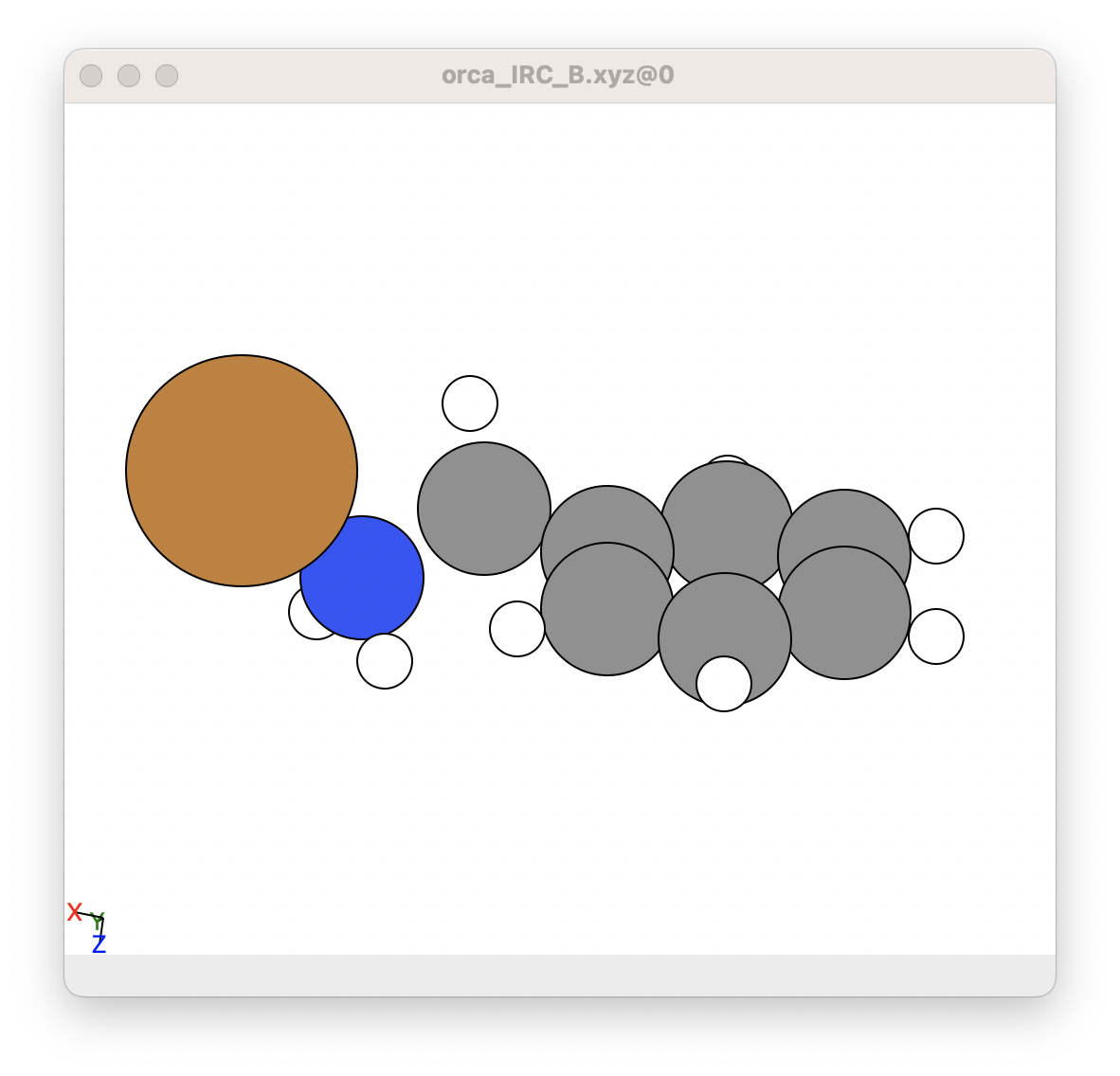
This is fine because the IRC is set by default with loose convergence criteria. These are the best settings for 4A, otherwise it takes forever to complete the IRC process. This is the reason for Step 4B: we then tighten the convergence with a regular geometric optimisation. By doing this, we get the following for the tightly converged backwards step.


We see that the tightly converged backwards structure from 4B gives a similar structure to the converged reactant from Step 1, so this is great.
One problem however is that the backwards structure from Step 4B did not converge. However, as we have mentioned this structure is very similar to the optimised reactant structure in Step 1.
- We could retry this calculation from the last geometric step from Step 4B, as it is common that ORCA will fail for various reasons and restarting ORCA sorts everything out
- However, because the backwards structure we got from Step 4B is pretty much the same as the optimised reactant from Step 1, I would be pragmatic and be happy that Step 4B backwards gave us the result we were looking for.
Troubleshooting the geometric optimisation (Opt) calculation¶
See the Troubleshooting Geometric Optimisation Calculations page for troubleshooting tips for geometric optimisations.