Step 2A - Prepare The Reaction System¶
Consider that you have a mechanistic step that contains two or more reactants/products. To perform Step 2 (either by performing a SCAN or Nudged Elastic Band calculation), you will need to place all your reactants/all your products into the same xyz file.
This section will look at how to append multiple chemical systems in various xyz files together so that you can examine the reaction pathway in either Step 2Ba - SCAN or Step 2Bb - Nudged Elastic Band.
Note
If your system only contains one structure where all atoms are bound to the same structure in some way, you do not need to do this step. Feel free to move on to Step 2Ba - SCAN or Step 2Bb - NEB.
Step 2A.1 - Move the Centre of Masses of your Chemical Systems¶
Before we combine multiple chemical systems together, we need to make sure that we don't accidentally place one molecule in the middle of another molecule. The easiest way to solve this problem is to move the centre of mass of your chemical systems some distance apart from each other. You can do this by using the viewORCA move_com module. To run this module:
- Change directory into the folder containing the
xyzfiles of the chemical systems you want to change the centre of masses of. - Run the
viewORCA move_com filepath --move_com_to=move_com_tomodule as shown below, wherefilepathis the path to thexyzfile you would like to move the centre of mass of, andmove_com_tois the \((x,y,z)\) coordinates where you would like to move the centre of mass of your system to.- Note: The format of
move_com_toshould bex,y,z. i.e:move_com_to=x,y,z
- Note: The format of
# First, change directory to the folder that contains the xyz files that you want to change the centre of masses of.
cd folder_containing_xyz_files
# Second, move the centre of mass of your chemical system
viewORCA move_com filepath --move_com_to=move_com_to
The viewORCA move_com module will create a new xyz file that contains move_com_to in the filename. This xyz file will contain your chemical system with its centre of mass moved to move_com_to.
Example
Suppose that we have a Cu(II)-Benzylimine that we want to react with Benzylamine. We decide that we want the Cu(II)-Benzylimine to be centred at the origin while we want Benzylamine to be centred at \((10.0, 0.0, 0.0)\). To do this we would do the following in the terminal:
# First, change directory to the folder that contains the Cu(II)-Benzylimine and Benzylamine xyz files.
cd folder_containing_xyz_Benzylamine.xyz
# Second, move the centre of mass of Cu(II)-Benzylimine to the origin (0,0,0).
viewORCA move_com Cu_II-BnzImine_1+.xyz --move_com_to=0,0,0
# Third, move the centre of mass of Benzylamine to (10,0,0).
viewORCA move_com Benzylamine.xyz --move_com_to=10,0,0
viewORCA move_com will move the centre of masses of Cu(II)-Benzylimine and Benzylamine and save them into the files called Cu_II-BnzImine_1+_com_0_0_0.xyz and Benzylamine_com_10.0_0.0_0.0.xyz. This example is given in Examples/Step2_Find_TS/Step_2A/Step_2A1_Move_COM.

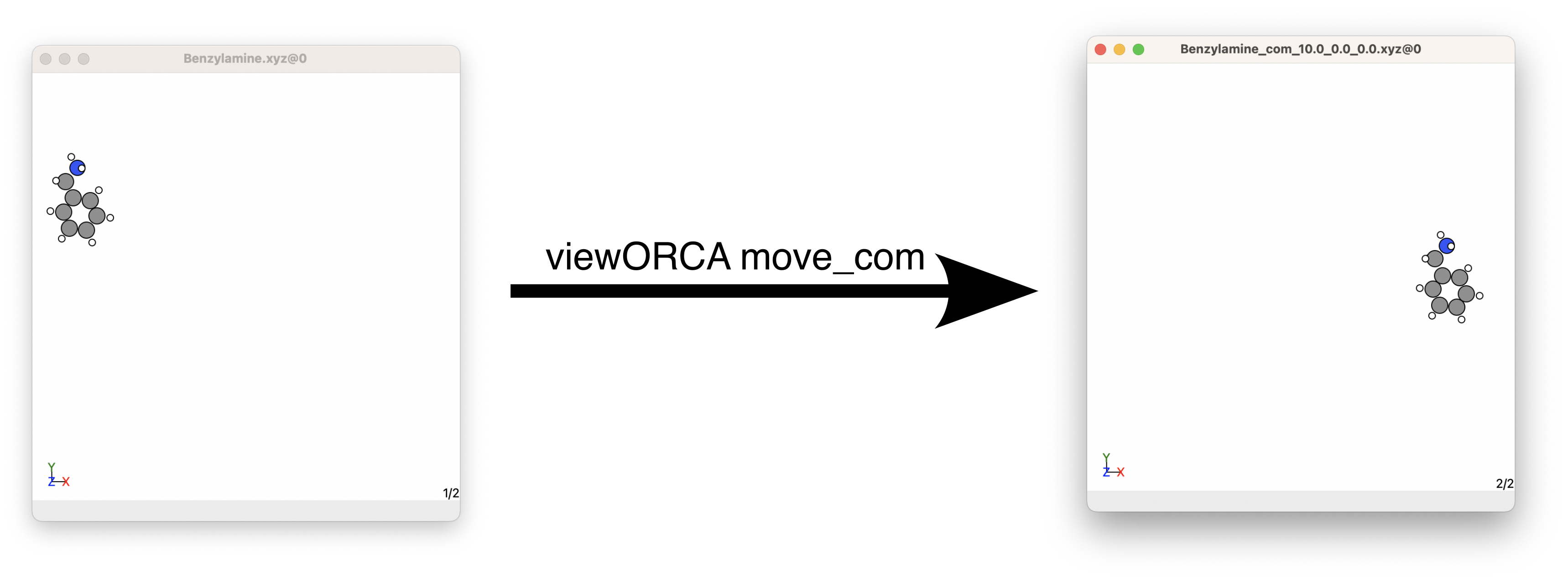
Step 2A.2 - Combine your xyz files into One Chemical System¶
Now that we have moved our molecules in Step 2A.1, we can now combine them together knowing that they won't accidentally merge together. There are two ways to do this:
Option 1: Use viewORCA combine¶
The first option is to use viewORCA combine to combine multiple xyz files together. To do this:
- In the terminal, change directory into the directory containing the xyz files of the chemical systems you want to combine together.
-
Type
viewORCA combine filepath_for_system_1 filepath_for_system_2 filepath_for_system_3 ...into the terminal, where:filepath_for_system_1, filepath_for_system_2, ...are the paths to all the xyz files you would like to combine together.
viewORCA combine will print the shortest distances between the chemical systems you combine together so you know they are a good distance from each other. If you see that any distance is less than 3.0 Å, you may have accidentally merged your chemical systems together.
The output file will be saved into your current working directory as overall_chemical_system.xyz. Check you have not accidentally merged your chemical systems by looking at the xyz file in a GUI (like Avogadro or ase gui). Once you are happy with the contents of this file, change the filename of this xyz file to what you want it to be named.
Example
Say we want to combine an xyz file containing a Cu(II)-benzylimine molecule (with centre of mass = \((0.0, 0.0, 0.0)\)) with another xyz file containing a benzylamine molecule (with centre of mass = \((10.0, 0.0, 0.0)\)). We would run the code below to achieve this:
# First, change directory into the folder containing your xyz files you want to combine together.
cd path_to_xyz_files
# Second, run "viewORCA combine".
viewORCA combine Benzylamine_com_10.0_0.0_0.0.xyz Cu_II-BnzImine_1+_com_0_0_0.xyz
viewORCA combine will print the shortest distances between the chemical systems you combine together so you know they are a good distance from each other. If you see that any distance is less than 3.0 Å, you may have accidentally merged your chemical systems together.
You will also see a new file has been made called overall_chemical_system.xyz that contains your overall chemical system. This example is given in Examples/Step2_Find_TS/Step_2A/Step_2A2_Combine.
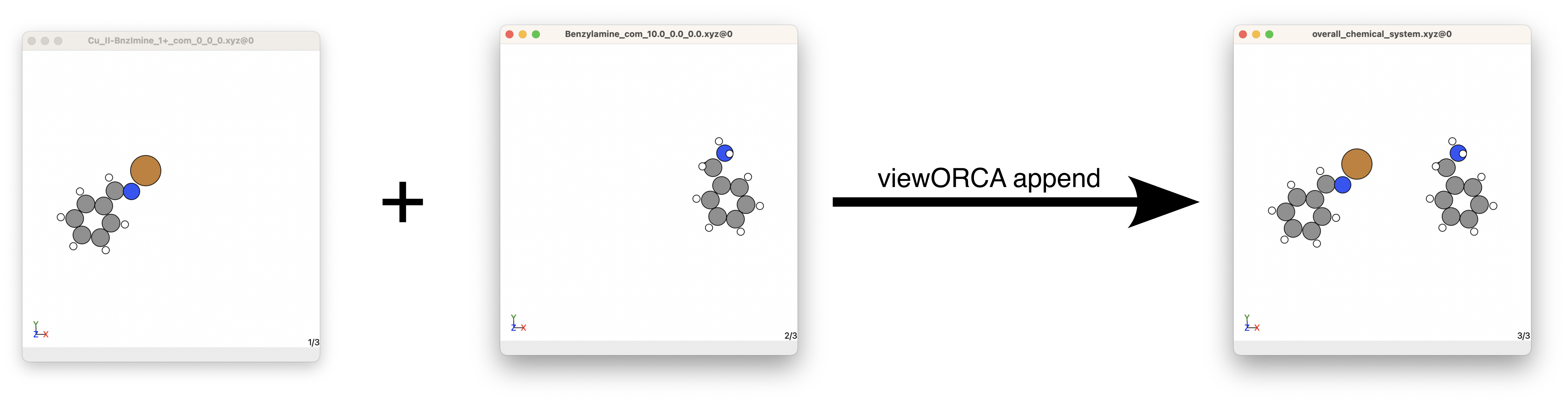
Option 2: Copy the coordinates of one xyz file into another xyz file¶
The other way to combine multiple xyz files together is to copy and paste each xyz file one after the other in a new xyz file. This is what is happening in Option 1: Use viewORCA combine, but done automatically by viewORCA.
- If you do this, make sure you change the first line in your
xyzfile to the total number of atoms in your combined system.
Example
Consider now that we want to manually combine the xyz file for Cu(II)-benzylimine molecule (with centre of mass = \((0.0, 0.0, 0.0)\)) with another xyz file containing a benzylamine molecule (with centre of mass = \((10.0, 0.0, 0.0)\)).
17
Properties=species:S:1:pos:R:3 Energy:=T 18.1285528=T pbc="F F F"
C 8.82798764 -0.72923719 -0.21980796
C 9.64720764 0.40132281 -0.05602796
C 9.37118764 -2.01621719 -0.16348796
C 10.73913764 -2.18955719 0.05608204
C 11.02826764 0.21373281 0.16385204
C 11.56615764 -1.07620719 0.21909204
H 12.62668764 -1.21243719 0.38803204
H 11.15850764 -3.18639719 0.09917204
H 11.69849764 1.05282281 0.29167204
H 7.76473764 -0.61273719 -0.39123796
H 8.73069764 -2.87946719 -0.29048796
C 9.00217764 1.76893281 -0.12848796
N 9.95429764 2.85972281 0.06699204
H 8.20434764 1.83886281 0.64416204
H 8.53106764 1.88010281 -1.12939796
H 10.30117764 2.83572281 1.05381204
H 9.44922764 3.76619281 -0.06206796
15
Properties=species:S:1:pos:R:3 Energy:=T 12.9406666=T pbc="F F F"
C -2.46950657 -0.54117045 -0.11252192
C -1.07283657 -0.69096045 -0.09875192
C -0.52041657 -1.97965045 -0.22782192
C -1.35452657 -3.09358045 -0.36770192
C -2.74232657 -2.93319045 -0.38011192
C -3.29917657 -1.65825045 -0.25265192
H -2.91808657 0.43999955 -0.01486192
H 0.55217343 -2.12735045 -0.22082192
H -0.92436657 -4.08190045 -0.46654192
H -3.38600657 -3.79657045 -0.48847192
H -4.37448657 -1.53509045 -0.26240192
C -0.21642657 0.50638955 0.05072808
N 1.06287343 0.41467955 0.06831808
H -0.67253657 1.48676955 0.14814808
Cu 2.15844343 2.02501955 0.26814808
To do this, we open a new file in our text editor (such as Sublime), and copy the elements and positions of all the atoms in Benzylamine_com_10.0_0.0_0.0.xyz, then do the same for Cu_II-BnzImine_1+_com_0_0_0.xyz.
At the top of this new file, we will need to add two lines:
- The first line indicates the number of atoms in this combined system, which in this case is
32 - The second line indicates what information about the atoms is given in this file (i.e., the element and \((x,y,z)\) position). This should be
Properties=species:S:1:pos:R:3 pbc="F F F"(pbchere indicates there is no periodic boundary condition set).
32
Properties=species:S:1:pos:R:3 pbc="F F F"
C 8.82798764 -0.72923719 -0.21980796
C 9.64720764 0.40132281 -0.05602796
C 9.37118764 -2.01621719 -0.16348796
C 10.73913764 -2.18955719 0.05608204
C 11.02826764 0.21373281 0.16385204
C 11.56615764 -1.07620719 0.21909204
H 12.62668764 -1.21243719 0.38803204
H 11.15850764 -3.18639719 0.09917204
H 11.69849764 1.05282281 0.29167204
H 7.76473764 -0.61273719 -0.39123796
H 8.73069764 -2.87946719 -0.29048796
C 9.00217764 1.76893281 -0.12848796
N 9.95429764 2.85972281 0.06699204
H 8.20434764 1.83886281 0.64416204
H 8.53106764 1.88010281 -1.12939796
H 10.30117764 2.83572281 1.05381204
H 9.44922764 3.76619281 -0.06206796
C -2.46950657 -0.54117045 -0.11252192
C -1.07283657 -0.69096045 -0.09875192
C -0.52041657 -1.97965045 -0.22782192
C -1.35452657 -3.09358045 -0.36770192
C -2.74232657 -2.93319045 -0.38011192
C -3.29917657 -1.65825045 -0.25265192
H -2.91808657 0.43999955 -0.01486192
H 0.55217343 -2.12735045 -0.22082192
H -0.92436657 -4.08190045 -0.46654192
H -3.38600657 -3.79657045 -0.48847192
H -4.37448657 -1.53509045 -0.26240192
C -0.21642657 0.50638955 0.05072808
N 1.06287343 0.41467955 0.06831808
H -0.67253657 1.48676955 0.14814808
Cu 2.15844343 2.02501955 0.26814808