Step 3: Optimise the Transition State¶
Read the theory first
Before starting this step, we recommend reading the theory for this step to understand why we are performing this step.
- Make sure you have also read the theory on the potential energy surface.
Tip
Click the Download Templates.zip button to download the template folder for this step.
- If you would like to follow this guide using data already calculated using ORCA, click
Download Examples.zip.
While your SCAN or NEB calculation will have done a good job at finding the transition state, it is a good idea to perform a transition state geometry optimisation to make sure that your transition state lies on the saddle point as best as possible.
To do this, we will optimise the transition state using the OptTS tag:
- The
OptTSmethod is designed to find the most optimal transition state by allowing the molecule to follow the eigenvector (vibrational mode) for the most negative eigenvalue (frequency).
To perform an OptTS calculation, we include the following line in the orca.inp:
The tags here indicate the following:
OptTS: Indicates that we want to optimise the molecule towards the transition state.NumFreq: We want to calculate the numerical frequency after the optimisation process to make sure that our transition state only has one negative vibrational frequency (transition states are defined as the lowest energy point between two local minima that has only 1 negative vibrational frequency).- We are using
NumFreqrather thanFreq(analytically calculated vibrational frequency) because of theGEOMsettings below (I think, based on the tutorial here)
- We are using
We also include the following in our inp file:
%GEOM
Calc_Hess true # Calculate Hessian in the beginning
NumHess true # Request numerical Hessian (analytical not available)
Recalc_Hess 1 # Recalculate the Hessian for every step
END
The reason for these settings is because we want to refine the transition state without converging towards a different saddle point unexpectedly. This can happen if the Hessian is slightly off, which can happen when using the Hessian approximation methods commonly used in computational chemistry applications.
- The Hessian describes the curvature (second order derivative) of the potential energy surface (PES). It is used to make a decision about how to modify your chemical system so you get closer to a converged system. It needs to be obtained for each geometric step you perform.
- Calculating the full Hessian is very computationally expensive, so approximations are commonly made so that the full Hessian does not need to be calculated after each geometric step.
- In this case, I have chosen to calculate the full Hessian after each step because I assume I am near the transition state after the SCAN/NEB calculation, and want to make sure it doesn't deviate from converging. This is probably overdoing it, however, and not completely necessary.
- From the tutorial here, they recalculate the full Hessian after 5 geometric steps rather than after every geometric step.
Note
See the tutorial here for a reference for these settings
Info
For all other ORCA calculations such as geometric optimisations, approximations to the Hessian are generally helpful and are recommended/set by default.
The full orca.inp file for this example is given below (also located here):
!B3LYP DEF2-TZVP D3BJ
!OptTS NumFreq TightOPT TightSCF defgrid2
%SCF
MaxIter 2000 # Here setting MaxIter to a very high number. Intended for systems that require sometimes 1000 iterations before converging (very rare).
DIISMaxEq 5 # Default value is 5. A value of 15-40 necessary for difficult systems.
directresetfreq 15 # Default value is 15. A value of 1 (very expensive) is sometimes required. A value between 1 and 15 may be more cost-effective.
END
%CPCM EPSILON 6.02 REFRAC 1.3723 END
%PAL NPROCS 32 END
%maxcore 2000
%GEOM
Calc_Hess true # Calculate Hessian in the beginning
NumHess true # Request numerical Hessian (analytical not available)
Recalc_Hess 1 # Recalculate the Hessian for every step
END
* xyzfile 1 1 orca.073.xyz
Outputs from ORCA¶
ORCA will create a number of files. These are the important ones to look at for optimising the transition state geometry:
output.out: The output file, which will tell you if the transition state converged or not.orca.xyz: This is the transition state found (if convergence was successful)orca_trj.xyz: This is the trajectory file that indicates the steps for how the calculation proceeded.
Once ORCA has finished, you want to do the following checks.
Check 1: Look at your transition state structure and the energy profile and make sure it looks ok¶
Just like in Step 1 for the reactants and products, you also want to look at your transition state and make sure it makes sense to you.
IMPORTANT
This is a very important check, as it is very possible that ORCA goes off track when locating the transition state. This is a problem not just for ORCA, but for any computational chemistry software.
You can check the transition state by opening up the orca.xyz file. You can do this in ASE by:
- Opening a new terminal
cdinto the optimisation folder- Type
ase gui orca.xyzinto the terminal
# cd into your transition state optimisation folder
cd ORCA_Mechanism_Procedure/Examples/Step3_Opt_TS
# View the transition state
ase gui orca.xyz
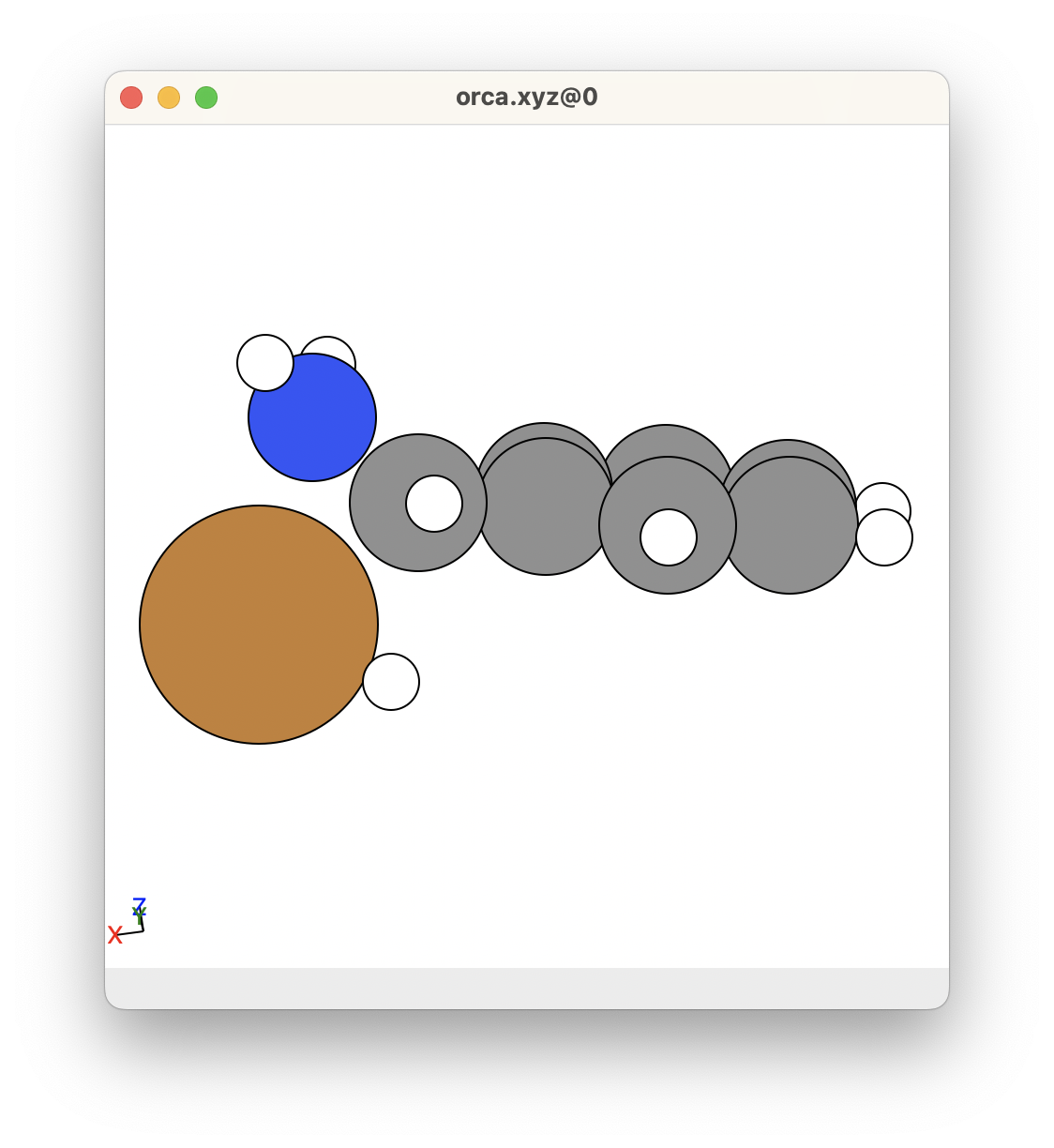
This is what I got for this example (see below).
- When comparing this transition state to the reactants and products, I can see that the hydrogen atom attached to the Cu atom has moved closer to the C atom.
- For this reason, I am happy with this transition state.
Check 2: Did the transition state converge successfully¶
Just like when we were checking convergence of the product and reactant in Step 1, we want to find the geometry convergence table from the ORCA output file and check that we are happy that the transition state converged properly, and we have got the HURRAY THE OPTIMIZATION HAS CONVERGED message:
.--------------------.
----------------------|Geometry convergence|-------------------------
Item value Tolerance Converged
---------------------------------------------------------------------
Energy change -0.0000006058 0.0000010000 YES
RMS gradient 0.0000068824 0.0000300000 YES
MAX gradient 0.0000245794 0.0001000000 YES
RMS step 0.0001699439 0.0006000000 YES
MAX step 0.0005685514 0.0010000000 YES
-------------------------------------------------------------------------
........................................................
Max(Bonds) 0.0001 Max(Angles) 0.02
Max(Dihed) 0.03 Max(Improp) 0.00
---------------------------------------------------------------------
***********************HURRAY********************
*** THE OPTIMIZATION HAS CONVERGED ***
*************************************************
Check 3: Check that we have only 1 negative vibrational frequency¶
We want to look at the frequency calculation results from the ORCA output file and check that there is only 1 negative frequency. This tells us that we are on a saddle point on the potential energy landscape (see the Theory page for why this works).
- Generally any negative frequency is good, but a negative frequency that is more negative than \(-100cm^{-1}\) is a good sign that the transition state is good.
- This is because the value of the frequency indicates the curvature of the transition state across the saddle point. The bigger the number, the steeper the energy descent on each side of the saddle point.
- A negative frequency between \(-20cm^{-1}\) and \(-100cm^{-1}\) is fine, but just beware you may have problems with Step 4 for technical reasons.
In the example below, you can see we only have one negative vibrational frequency, so our transition state is good to go!
-----------------------
VIBRATIONAL FREQUENCIES
-----------------------
Scaling factor for frequencies = 1.000000000 (already applied!)
0: 0.00 cm**-1
1: 0.00 cm**-1
2: 0.00 cm**-1
3: 0.00 cm**-1
4: 0.00 cm**-1
5: 0.00 cm**-1
6: -822.13 cm**-1 ***imaginary mode***
7: 58.18 cm**-1
8: 84.13 cm**-1
9: 171.84 cm**-1
10: 228.59 cm**-1
11: 260.60 cm**-1
12: 361.45 cm**-1
13: 413.26 cm**-1
14: 427.65 cm**-1
15: 490.08 cm**-1
16: 523.52 cm**-1
17: 579.22 cm**-1
18: 633.50 cm**-1
19: 639.19 cm**-1
20: 695.96 cm**-1
21: 713.56 cm**-1
22: 792.39 cm**-1
23: 809.13 cm**-1
24: 861.16 cm**-1
25: 949.18 cm**-1
26: 991.84 cm**-1
27: 1017.62 cm**-1
28: 1022.53 cm**-1
29: 1049.37 cm**-1
30: 1092.26 cm**-1
31: 1104.90 cm**-1
32: 1144.41 cm**-1
33: 1181.20 cm**-1
34: 1182.34 cm**-1
35: 1208.39 cm**-1
36: 1244.74 cm**-1
37: 1336.79 cm**-1
38: 1369.30 cm**-1
39: 1437.49 cm**-1
40: 1492.70 cm**-1
41: 1532.23 cm**-1
42: 1618.01 cm**-1
43: 1621.65 cm**-1
44: 1641.40 cm**-1
45: 1776.88 cm**-1
46: 3170.21 cm**-1
47: 3174.89 cm**-1
48: 3183.78 cm**-1
49: 3191.79 cm**-1
50: 3197.99 cm**-1
51: 3200.88 cm**-1
52: 3527.19 cm**-1
53: 3629.01 cm**-1
Check 4 (Optional): Check how ORCA optimised your transition state¶
If you think there might be something funny happening, it is sometimes a good idea to check how ORCA optimised your transition state.
You can do this by changing directory into the transition state folder and typing viewORCA opt into the terminal.
# change directory into the Step3_Opt_TS folder
cd ORCA_Mechanism_Procedure/Examples/Step3_Opt_TS
# View the geometry optimisation by ORCA in ASE using viewORCA opt
viewORCA opt
NOTE 1
viewORCA opt will also create a xyz file called OPT_images.xyz that you can copy and view on your own computer.
- If you just want to create the
OPT_images.xyzfile, type into the terminalviewORCA opt --view False(which will create theOPT_images.xyzfile without opening anase guiwindow).
NOTE 2
Do not expect the energy to go down. The energy may go up during the geometry optimisation, as we are trying to find a saddle point on the potential energy surface rather than a local minimum.
Other Information about performing transition state optimisations in ORCA¶
Click here to learn more about transition state calculations from the ORCA Input Library.
Note
The ORCA Input Library is a great source of information about performing calculations in ORCA.
Troubleshooting transition state geometry optimisations (OptTS) calculations¶
Here are some troubleshooting tips for performing this optimisation step.
No troubleshooting issues have been found yet.
Info
If you have any issues about this step, raise a New issue at the Issues section by clicking here.