viewORCA Manual¶
There are several modules available for viewing ORCA calculations. This webpage describes all the modules available in viewORCA:
Info
You can view all the module available in viewORCA by typing viewORCA --help into the terminal:
username@computer name Desktop % viewORCA --help
usage: viewORCA [-h] [-T] {help,opt,scan,neb,neb_snap,irc,multi,move_com,combine} ...
Program for viewing jobs from ORCA
optional arguments:
-h, --help show this help message and exit
-T, --traceback
Sub-commands:
{help,opt,scan,neb,neb_snap,irc,multi,move_com,combine}
help Help for sub-command.
opt This module allows the user to view the output of an ORCA geometric optimisation job visually.
scan This module allows the user to view the output of an ORCA SCAN job visually.
neb This module allows the user to view the output of an ORCA Nudged Elastic Band (NEB) job visually.
neb_snap This module allows the user to view the progress of an ORCA Nudged Elastic Band (NEB) job (as it is running or after it has run) by viewing the
energy profiles of all the iterations of the NEB optimisation job.
irc This module allows the user to view the output of an ORCA Intrinsic Reaction Coordinate (IRC) job visually.
multi This module allows the user to obtain the number of electrons in the system of interest, and therefore obtain the possible multiplicities that the
system could be in.
move_com This module allows the user to move the centre of mass of a system.
combine This module allows the user to combine a set of chemical systems together.
viewORCA opt: View Geometric Optimisation Calculation¶
This module is designed to allow the user to view the output of an ORCA geometric optimisation job visually.
To use this module:
- In the terminal, change directory into the directory containing your geometric optimisation job, and
- type
viewORCA optinto the terminal.
# First, change directory into the folder containing your ORCA geometric optimisation job.
cd path_to_geometric_optimisation_job
# Second, run "viewORCA opt".
viewORCA opt
viewORCA opt will extract information from the ORCA_INPUT_FILENAME_trj.xyz file and show the images of the optimisation, along with the energy profile of the optimisation.
- For this reason, you MUST have the
ORCA_INPUT_FILENAME_trj.xyzfile in your geometric optimisation folder forviewORCA optto work. - The prefix
ORCA_INPUT_FILENAMEis the same prefix you gave to your ORCA input file (beingORCA_INPUT_FILENAME.inp).
This program will also create an xyz file called ORCA_INPUT_FILENAME_OPT_images.xyz that contains the images and the energy profile from the optimisation.
-
You can view this by typing
ase gui ORCA_INPUT_FILENAME_OPT_images.xyzinto the terminal.
Optional inputs for viewORCA opt include:
--path: This is the path to the folder containing the ORCA optimisation calculation. If not given, the default path is the current directory you are in.--view: If tag indicates if you want to view the ASE GUI for the optimisation job immediate after this program has run. By default this isTrue. If you only want to create theORCA_INPUT_FILENAME_OPT_images.xyzfile and not view it immediately, set this toFalse.- Setting this to
Falseis ideal if you are runningviewORCAon an HPC, wherease guiwindows may be more responsive ifase guiis run from your own computer rather than directly on the HPC. - You can view
ORCA_INPUT_FILENAME_OPT_images.xyzon your computer by typing the following into the terminal:ase gui ORCA_INPUT_FILENAME_OPT_images.xyz.
- Setting this to
Example
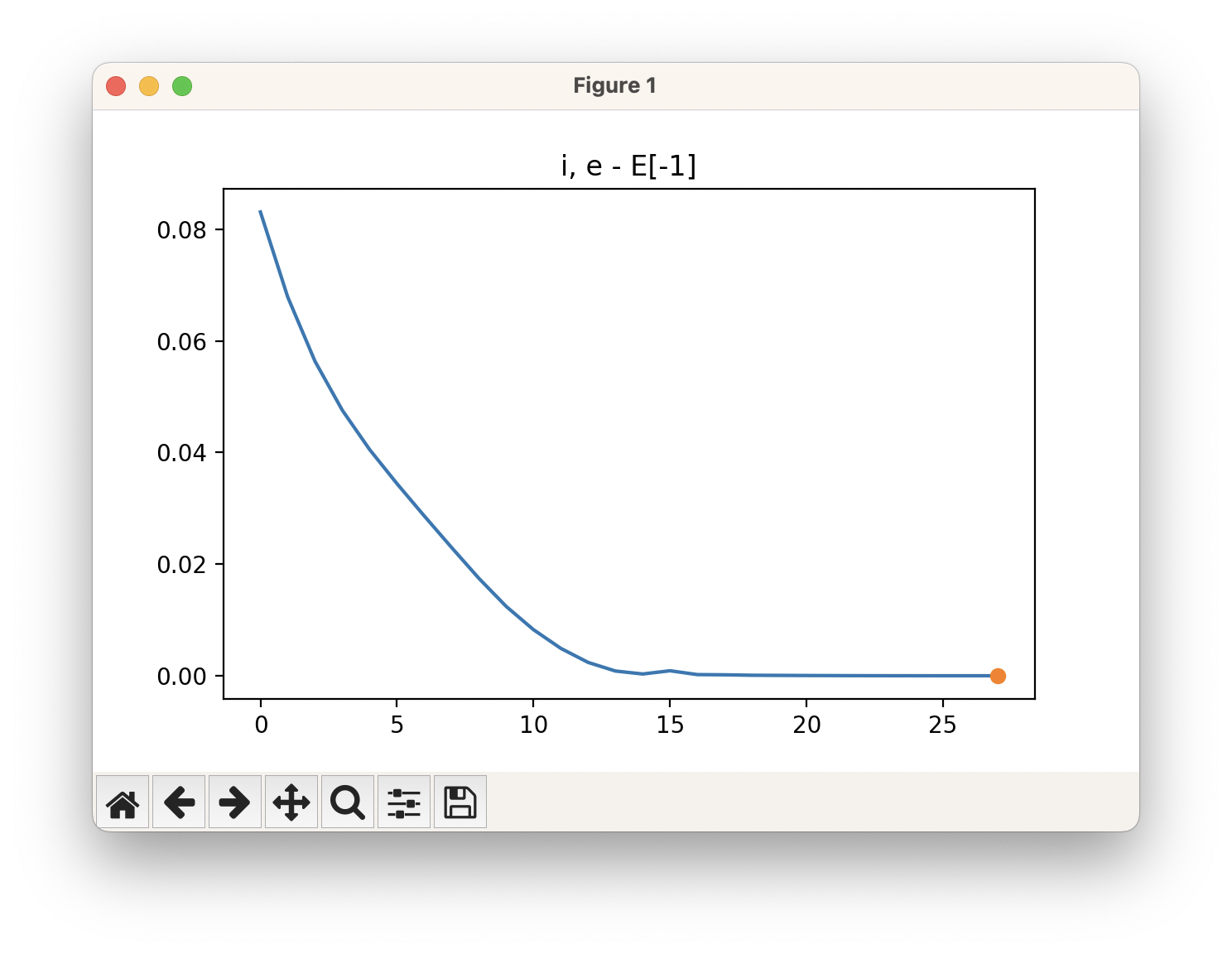
An example of a geometric optimisation viewed using viewORCA opt is shown below, along with the energy profile for this optimisation. This example is given in Examples/viewORCA_opt
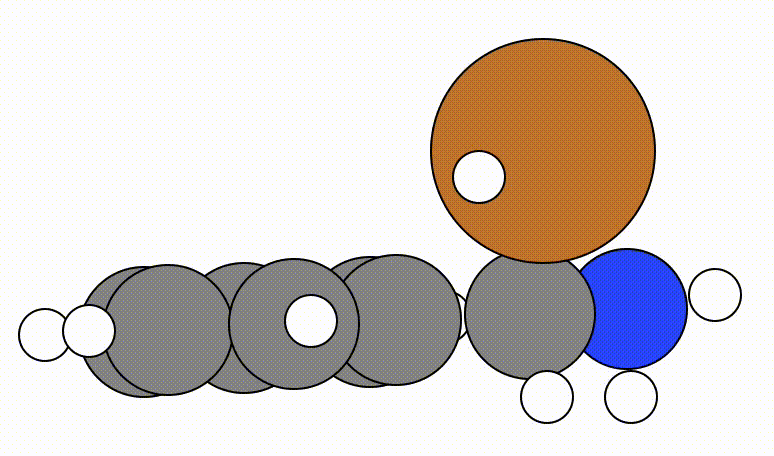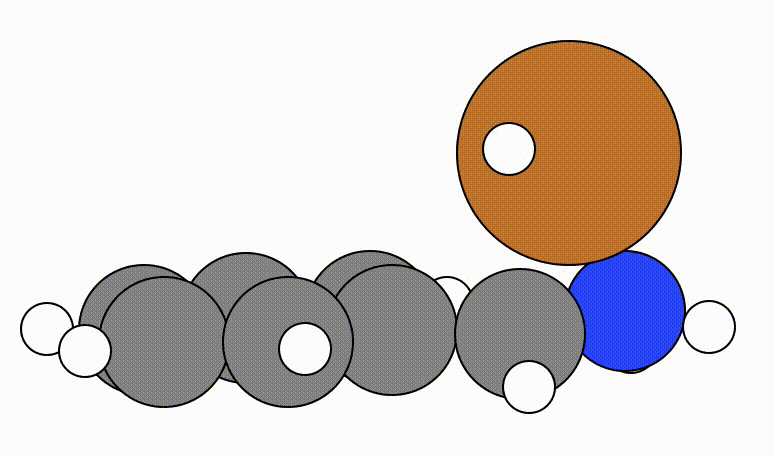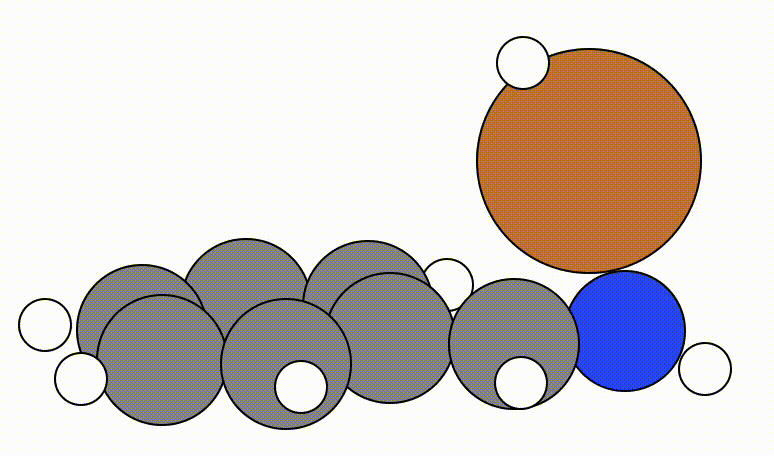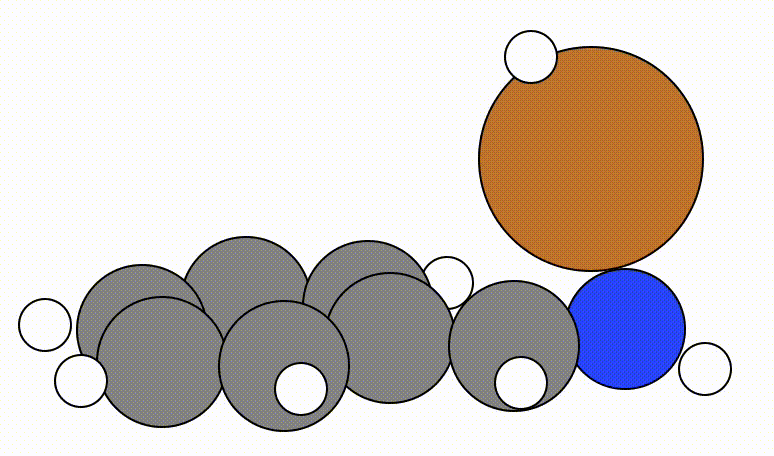
The energy profile for this example is given below:
viewORCA scan: View SCAN Calculation¶
This module is designed to allow the user to view the output of an ORCA SCAN job visually.
To use this module:
- In the terminal, change directory into the directory containing your SCAN job, and
- type
viewORCA scaninto the terminal.
# First, change directory into the folder containing your ORCA SCAN job.
cd path_to_SCAN_job
# Second, run "viewORCA scan".
viewORCA scan
Note
If you needed to restart your SCAN calculation part-way though the calculation for some reason and have multiple SCAN calculations that consectively run one after the other, you can run these sets of calculations together through viewORCA scan
viewORCA scanwill cellotape these calculations together so you can see the SCAN images together, and the full energy profile in one plot.
To use viewORCA scan in this way, make sure that your SCAN jobs are placed in separate folders called SCAN_1, SCAN_2, SCAN_3, so on and so on. viewORCA scan will paste these together in numerical order of these folders.
viewORCA scan will show the images of the SCAN calculation, along with the energy profile of the SCAN calculation.
-
This program will also create an xyz file called
SCAN_images.xyzthat contains the images and the energy profile from the SCAN calculation. You can view this by typingase gui SCAN_images.xyzinto the terminal.
Optional inputs for viewORCA scan include:
--path: This is the path to the folder containing the ORCA SCAN calculation. If not given, the default path is the current directory you are in.--view: If tag indicates if you want to view the ASE GUI for the SCAN job immediate after this program has run. By default this isTrue. If you only want to create theSCAN_images.xyzfile and not view it immediately, set this toFalse.- Setting this to
Falseis ideal if you are runningviewORCAon an HPC, wherease guiwindows may be more responsive ifase guiis run from your own computer rather than directly on the HPC. - You can view
SCAN_images.xyzon your computer by typing the following into the terminal:ase gui SCAN_images.xyz.
- Setting this to
Example
An example of a SCAN calculation viewed using viewORCA scan is shown below, along with the energy profile for this SCAN calculation. This example is given in Examples/viewORCA_scan
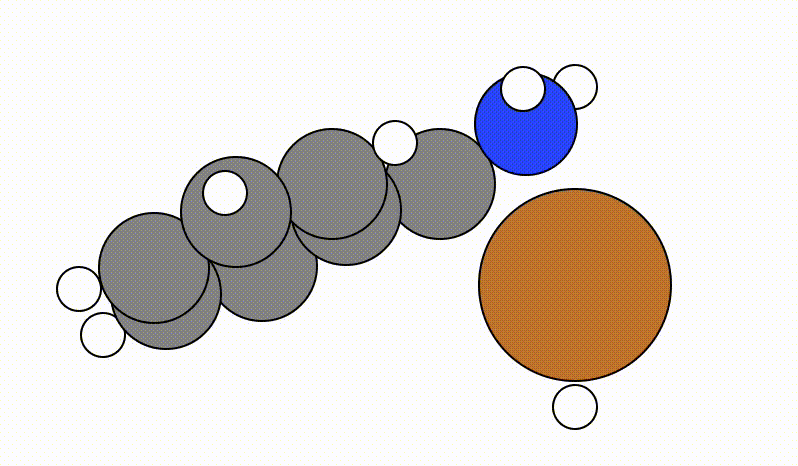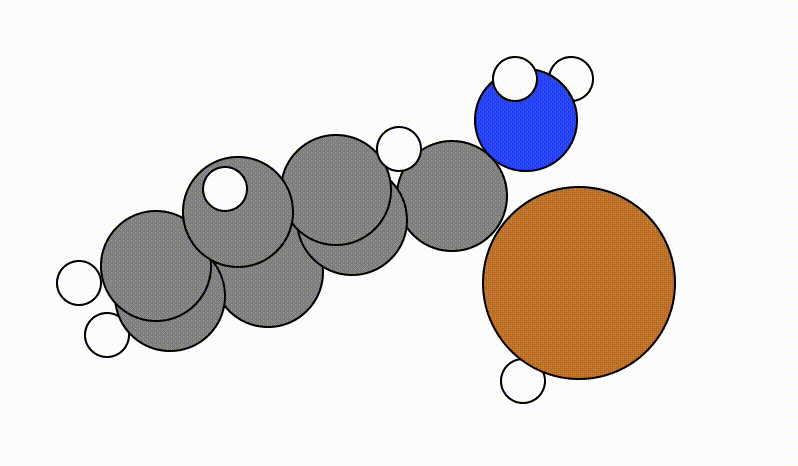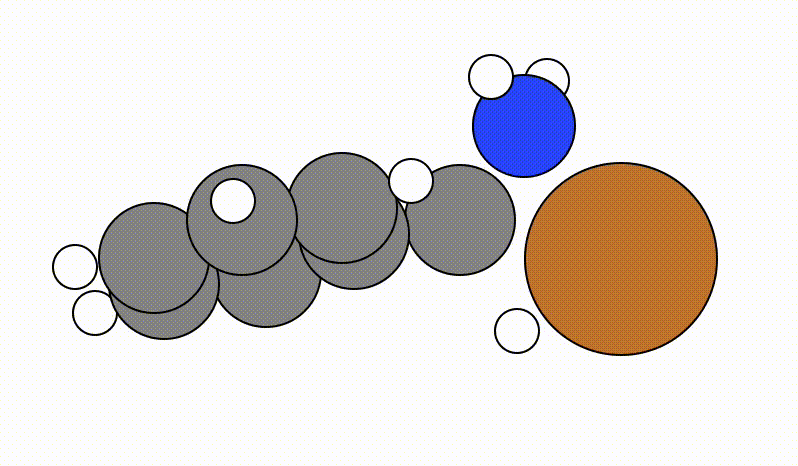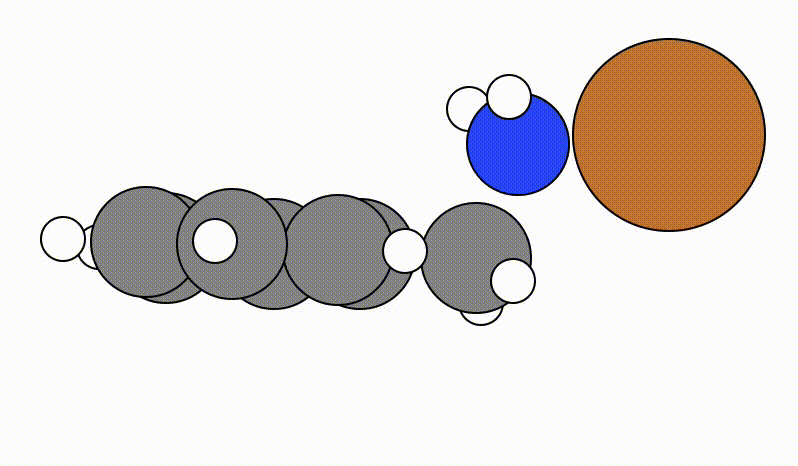
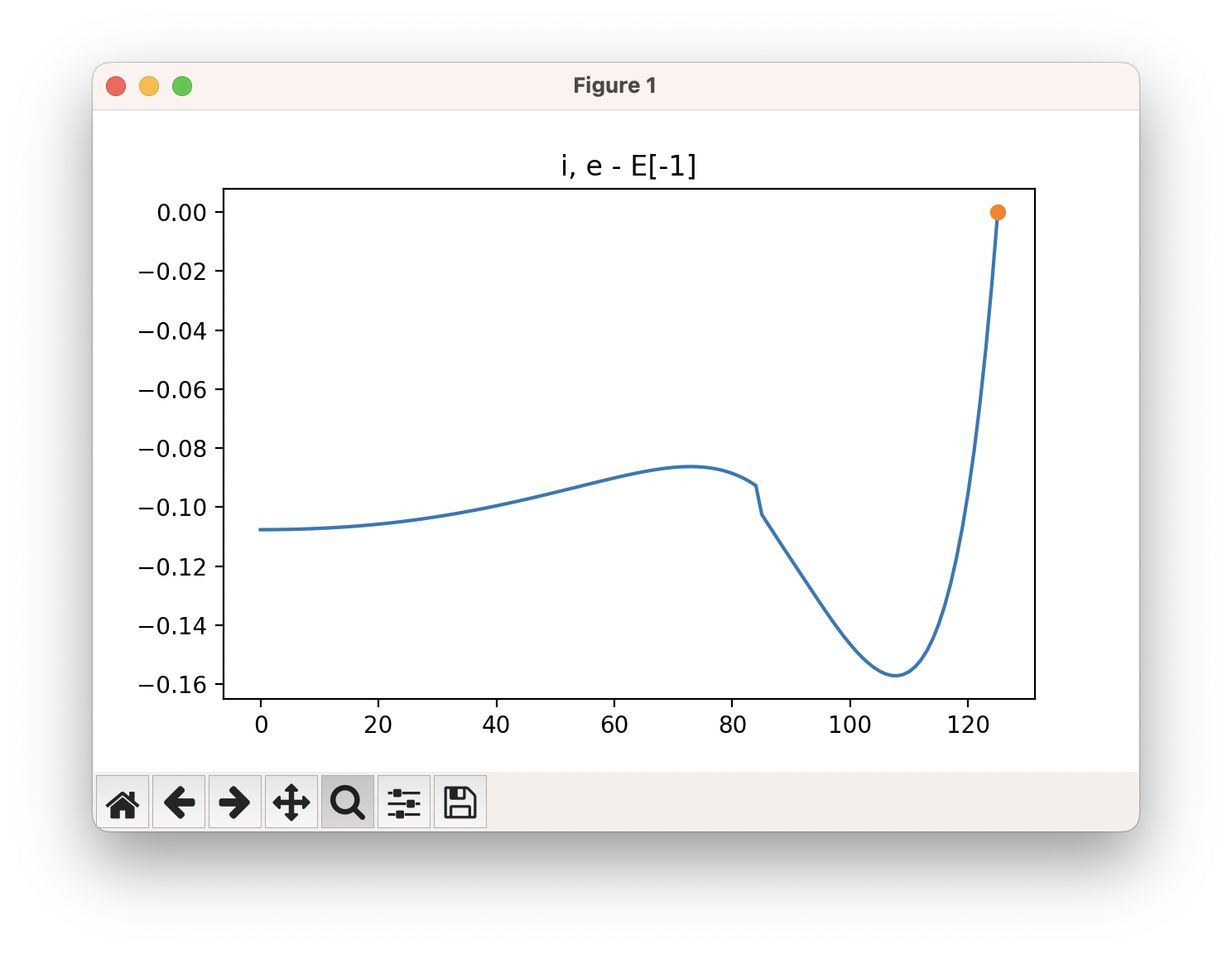
The energy profile for this example is given below:
viewORCA neb: View Nudged Elastic Band (NEB) Calculation¶
This module is designed to allow the user to view the output of an ORCA NEB job visually.
To use this module:
- In the terminal, change directory into the directory containing your NEB job, and
- type
viewORCA nebinto the terminal.
# First, change directory into the folder containing your ORCA NEB job.
cd path_to_NEB_job
# Second, run "viewORCA neb".
viewORCA neb
viewORCA neb will extract information from the ORCA_INPUT_FILENAME_MEP_trj.xyz file and show the images of the NEB calculation, along with the energy profile of the NEB calculation.
- For this reason, you MUST have the
ORCA_INPUT_FILENAME_MEP_trj.xyzfile in your NEB calculation folder forviewORCA nebto work. - The prefix
ORCA_INPUT_FILENAMEis the same prefix you gave to your ORCA input file (beingORCA_INPUT_FILENAME.inp).
This program will also create an xyz file called ORCA_INPUT_FILENAME_NEB_images.xyz that contains the images and the energy profile from the NEB calculation.
-
You can view this by typing
ase gui ORCA_INPUT_FILENAME_NEB_images.xyzinto the terminal.
Optional inputs for viewORCA neb include:
--path: This is the path to the folder containing the ORCA NEB calculation. If not given, the default path is the current directory you are in.--view: If tag indicates if you want to view the ASE GUI for the NEB job immediate after this program has run. By default this isTrue. If you only want to create theORCA_INPUT_FILENAME_NEB_images.xyzfile and not view it immediately, set this toFalse.- Setting this to
Falseis ideal if you are runningviewORCAon an HPC, wherease guiwindows may be more responsive ifase guiis run from your own computer rather than directly on the HPC. - You can view
ORCA_INPUT_FILENAME_NEB_images.xyzon your computer by typing the following into the terminal:ase gui ORCA_INPUT_FILENAME_NEB_images.xyz.
- Setting this to
Example
An example of a NEB calculation viewed using viewORCA neb is shown below, along with the energy profile for this NEB calculation. This example is given in Examples/viewORCA_neb_and_neb_snap
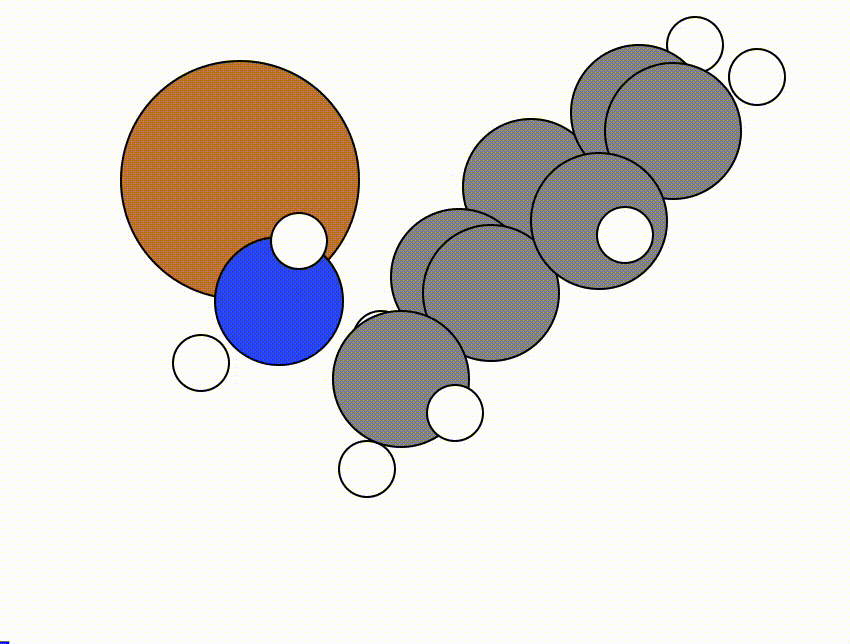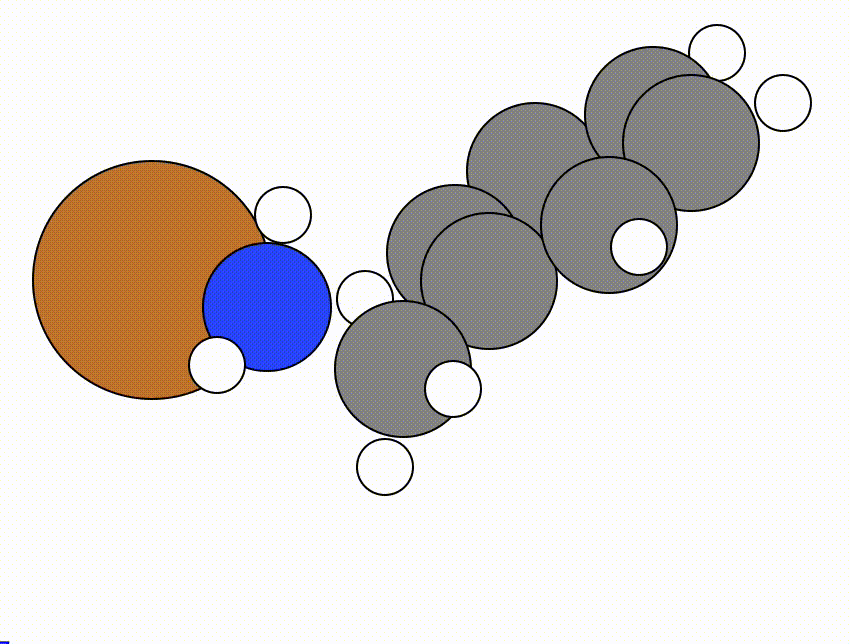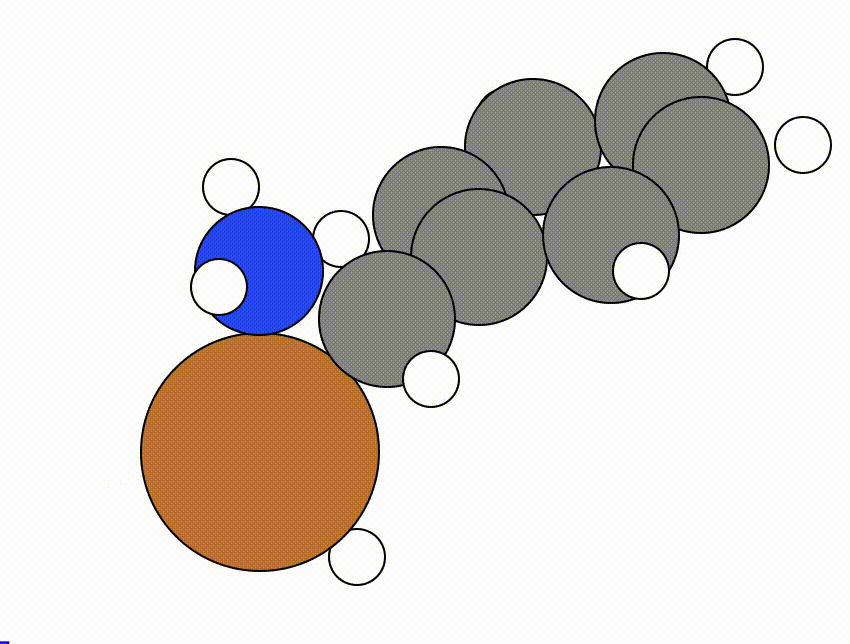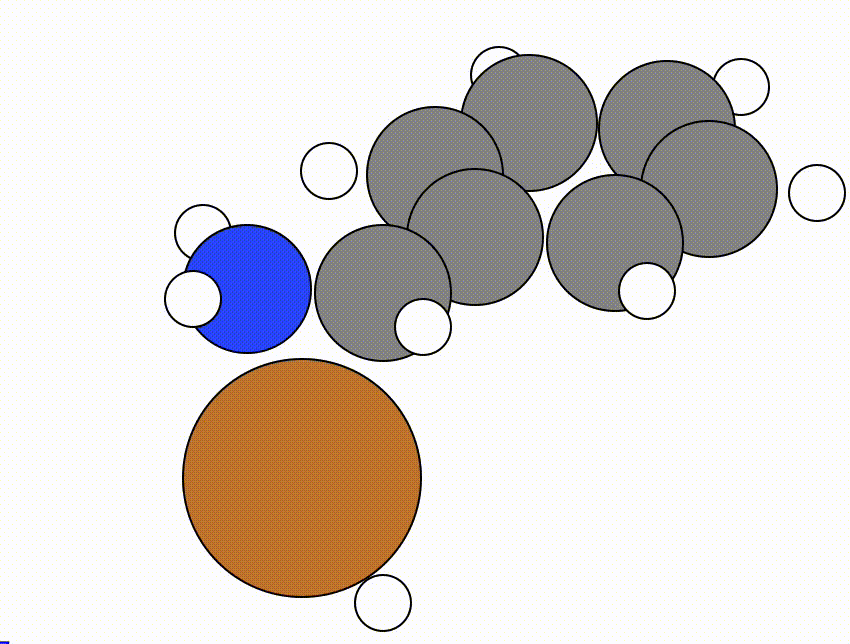
The energy profile for this example is given below:
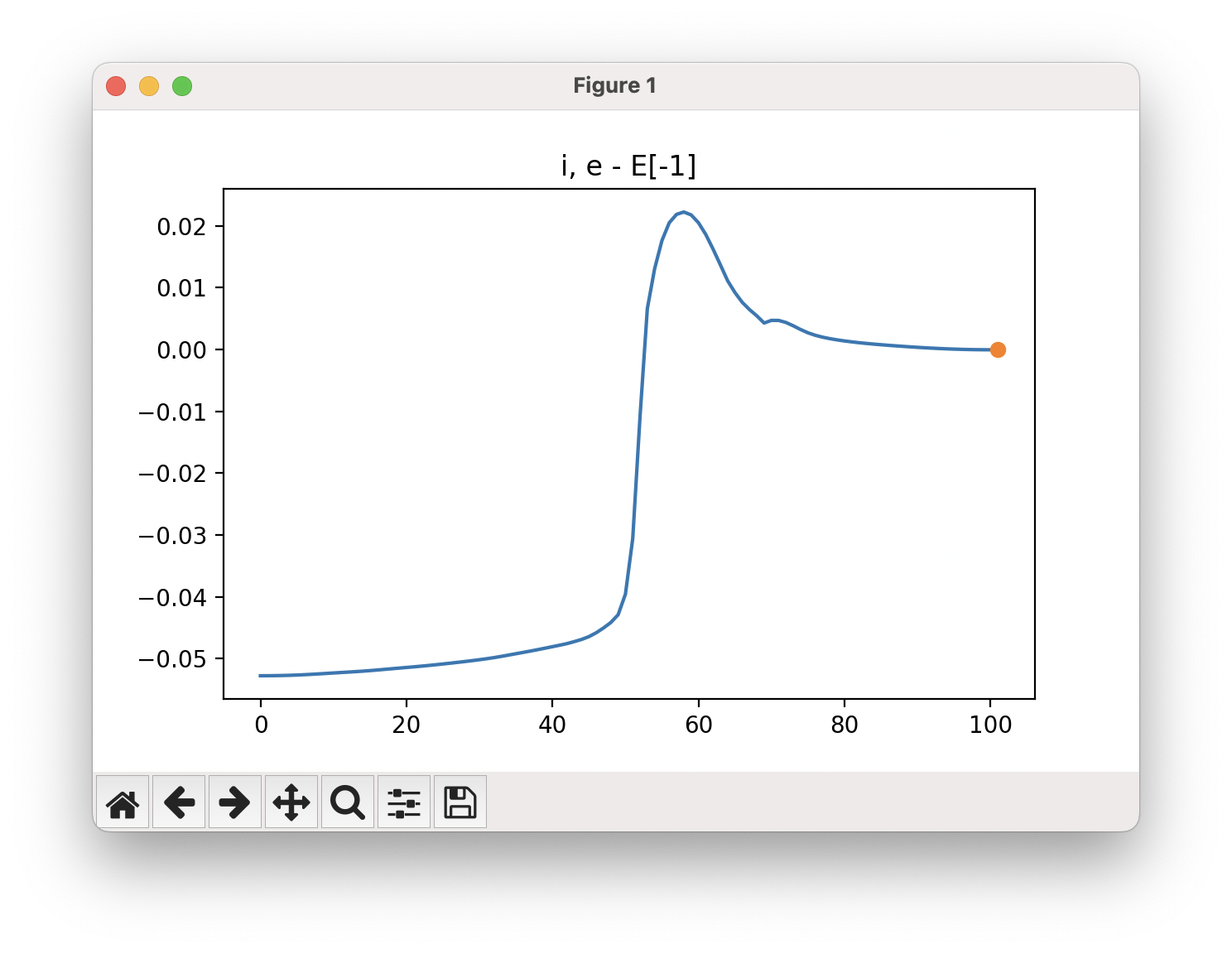
viewORCA neb_snap: View NEB Optimisation Process¶
This module is designed to allow the user to view the energy profile of all the iterations performed during the NEB optimisation process. This module is an adaptation of neb_snapshots.py by Vilhjálmur Ásgeirsson and Benedikt Orri Birgirsson of the Universiy of Iceland.
- It is helpful to view this as your NEB calculation is running to check if there are any issues occurring as ORCA us running the NEB calculation.
To use this module:
- In the terminal, change directory into the directory containing your NEB job, and
- type
viewORCA neb_snapinto the terminal.
# First, change directory into the folder containing your ORCA NEB job.
cd path_to_NEB_job
# Second, run "viewORCA neb_snap".
viewORCA neb_snap
viewORCA neb_snap will extract information from the ORCA_INPUT_FILENAME.interp file and create plots of the energy profiles of all the iterations of the NEB optimisation process.
- For this reason, you MUST have the
ORCA_INPUT_FILENAME.interpfile in your NEB calculation folder forviewORCA neb_snapto work. - The prefix
ORCA_INPUT_FILENAMEis the same prefix you gave to your ORCA input file (beingORCA_INPUT_FILENAME.inp).
viewORCA neb_snap will create two png files containing the energy profiles of the NEB calculation:
ORCA_INPUT_FILENAME_NEB_optimization.png: This plot displays the NEB energy profiles of all iterations performed during the NEB optimisation process.ORCA_INPUT_FILENAME_NEB_last_iteration.png: This plot shows the energy profile of the last iteration performed from the NEB optimisation process.
Optional inputs for viewORCA neb_snap include:
--path: This is the path to the folder containing the ORCA NEB calculation. If not given, the default path is the current directory you are in.- You can also provide the path to the
.interpfile you want to process if you want to process a specific.interpfile of interest. This file must end with the.interpsuffix.
- You can also provide the path to the
Example
An example of a NEB optimisation calculation viewed using viewORCA neb_snap is shown below, along with the energy profile of the last iteration for this NEB calculation. This example is given in Examples/viewORCA_neb_and_neb_snap
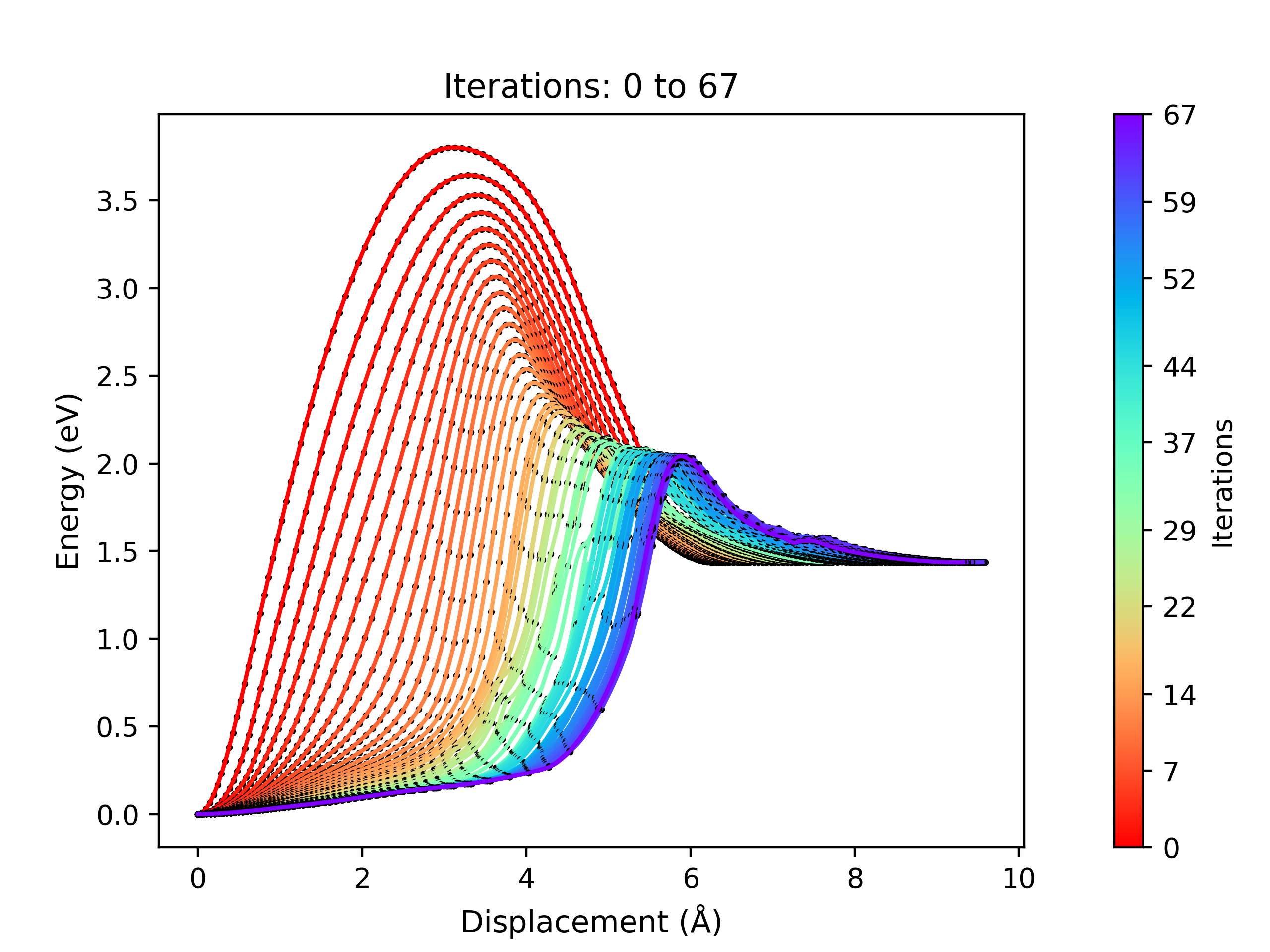
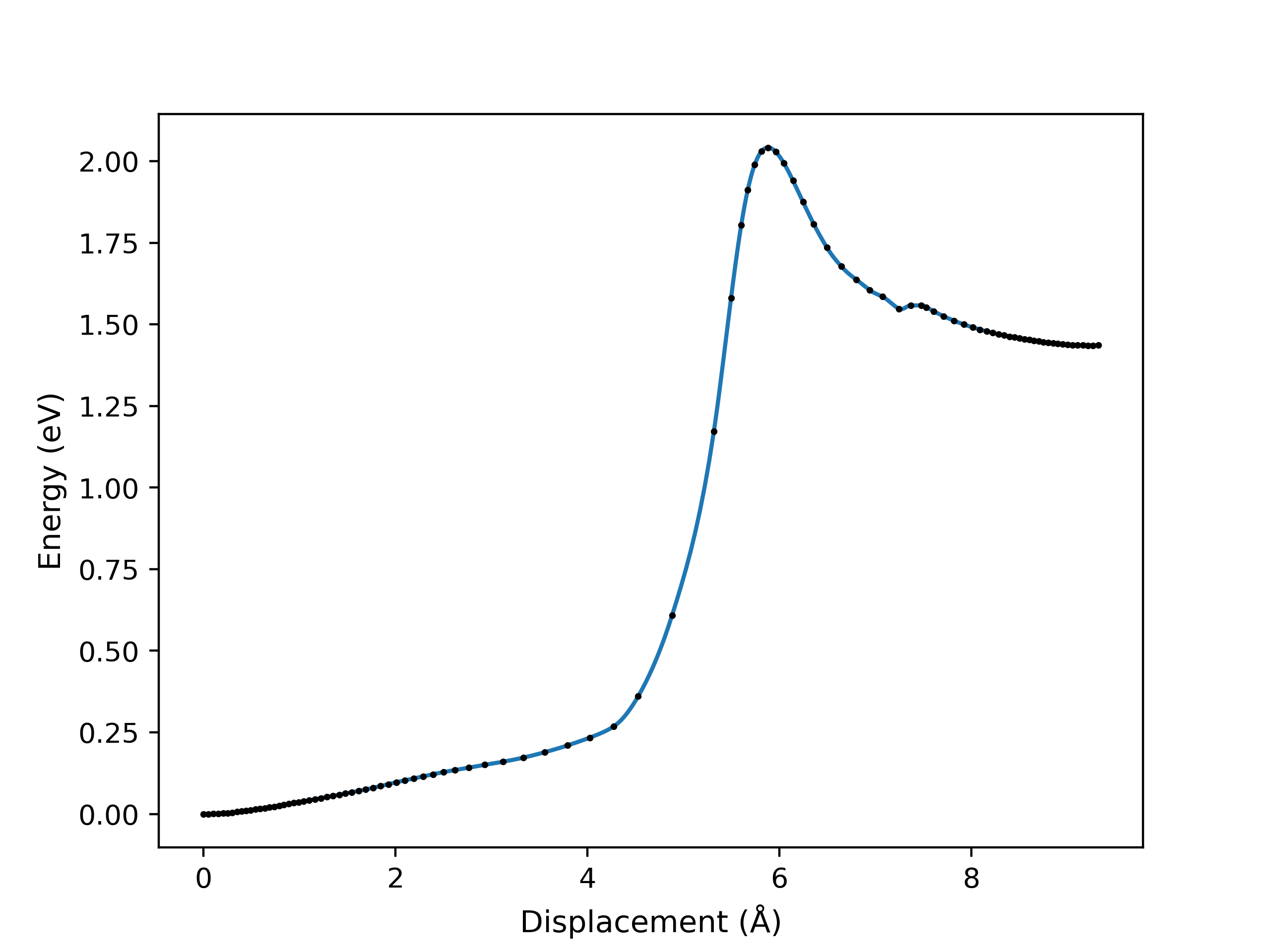
viewORCA irc: View Intrinsic Reaction Coordinate (IRC) Calculation¶
This module is designed to allow the user to view the output of an ORCA IRC job visually.
To use this module:
- In the terminal, change directory into the directory containing your IRC job, and
- type
viewORCA ircinto the terminal.
# First, change directory into the folder containing your ORCA IRC job.
cd path_to_geometric_optimisation_job
# Second, run "viewORCA irc".
viewORCA irc
viewORCA irc will extract information from the ORCA_INPUT_FILENAME_IRC_Full_trj.xyz file and show the images of the IRC calculation, along with the energy profile of the IRC calculation.
- For this reason, you MUST have the
ORCA_INPUT_FILENAME_IRC_Full_trj.xyzfile in your NEB calculation folder forviewORCA nebto work. - The prefix
ORCA_INPUT_FILENAMEis the same prefix you gave to your ORCA input file (beingORCA_INPUT_FILENAME.inp).
This program will also create an xyz file called ORCA_INPUT_FILENAME_IRC_images.xyz that contains the images and the energy profile from the IRC calculation.
-
You can view this by typing
ase gui ORCA_INPUT_FILENAME_IRC_images.xyzinto the terminal
Optional inputs for viewORCA irc include:
--path: This is the path to the folder containing the ORCA IRC calculation. If not given, the default path is the current directory you are in.--view: If tag indicates if you want to view the ASE GUI for the IRC job immediate after this program has run. By default this isTrue. If you only want to create theSCAN_images.xyzfile and not view it immediately, set this toFalse.- Setting this to
Falseis ideal if you are runningviewORCAon an HPC, wherease guiwindows may be more responsive ifase guiis run from your own computer rather than directly on the HPC. - You can view
ORCA_INPUT_FILENAME_IRC_images.xyzon your computer by typing the following into the terminal:ase gui ORCA_INPUT_FILENAME_IRC_images.xyz.
- Setting this to
Example
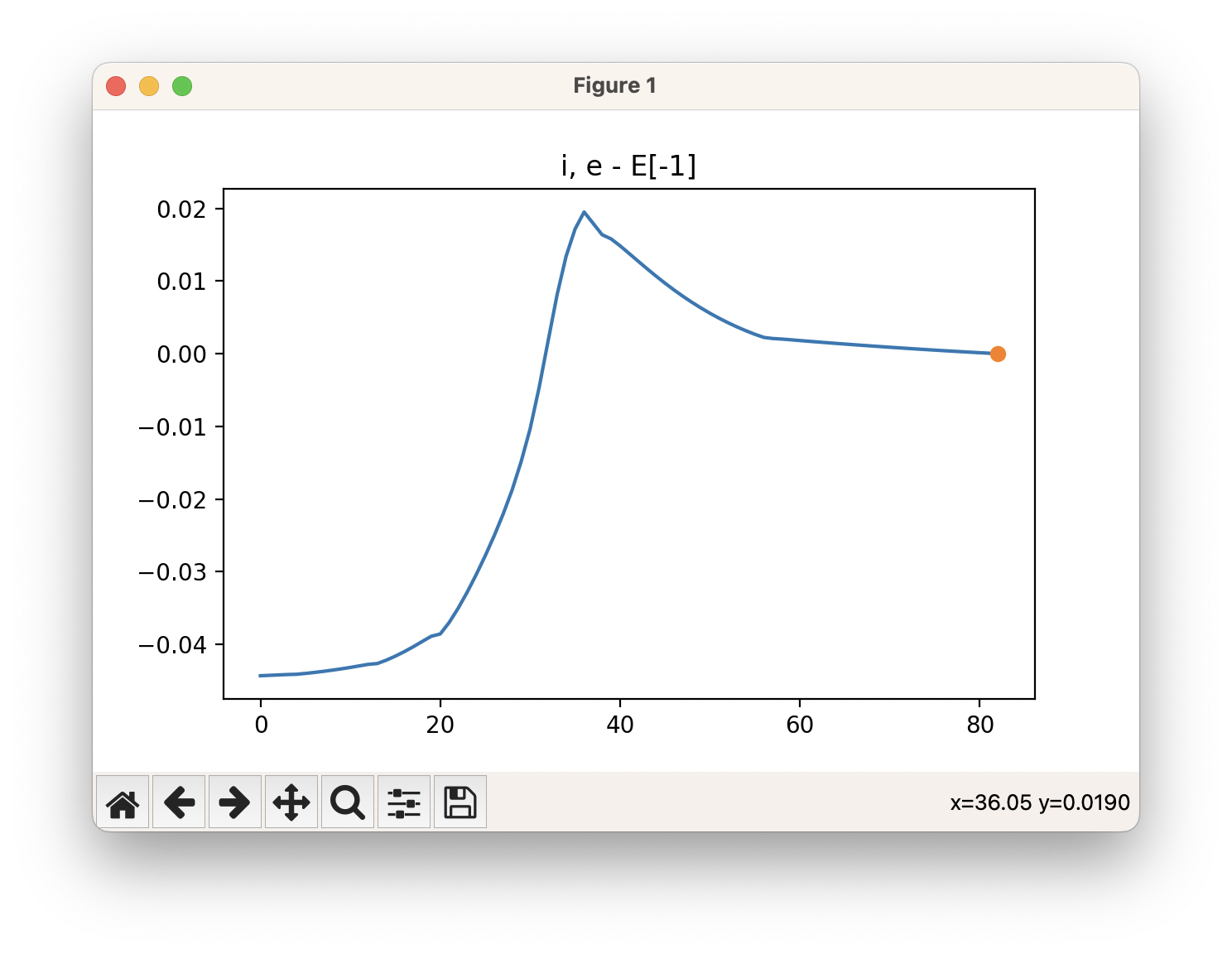
An example of a IRC calculation viewed using viewORCA irc is shown below, along with the energy profile for this IRC calculation. This example is given in Examples/viewORCA_irc
The energy profile for this example is given below:
viewORCA multi: Obtain Possible Electron Multiplicities for your System¶
One of the inputs that you need to give to your ORCA input .inp file is the multiplicity of the system. The multiplicity indicates the total electron spin of your system. The value of the muliticity is based on the number of electrons are in your system, where the multiplicity is based on the \(M = 2S + 1\) rule:
- If you have an even number of electrons in your system, the multiplicity will be an odd number. The lowest value multiplicity is 1.
- If you have an odd number of electrons in your system, the multiplicity will be an even number. The lowest value multiplicity is 2 (It can not be 0).
If you want to easily figure out if you should have an even or odd multiplicity, the viewORCA multi will determine the number of electrons your system has, and from this indicate if your multiplicity will be even or odd.
To use this module:
- In the terminal, change directory into the directory containing your geometric optimisation job, and
-
type
viewORCA multi filepath --charge=chargeinto the terminal, where:filepathis the path to thexyzfile of your system, and--chargeis the overall charge of your system (if--chargeis not given, the default given is 0).
Example 1: Cu(I)-Benzylamine
Cu(II)-Benzylimion (with a missing hydrogen and having a charge of +1) contains 86 electrons, so it can have an odd multiplicity. For Cu(I) systems, the ground spin state multiplicity is 1. See the xyz file for Cu(II)-Benzylimion in Examples/viewORCA_multi/Example_1
username@computer:username/Desktop$ viewORCA multi Cu_I-Benzylamine.xyz --charge=1
Number of electrons in Cu_I-Benzylamine.xyz is: 86
The total number of electrons in the system is even.
The multiplicity can be an odd number greater than or equal to 1 based on the M=2S+1 rule. Examples: 1,3,5,7,9,11,..


Example 2: Cu(II)-Benzylimion
Cu(II)-Benzylimion (with a missing hydrogen and having a charge of +1) contains 83 electrons, so it can have an even multiplicity. For Cu(II) systems, the ground spin state multiplicity is 2. See the xyz file for Cu(II)-Benzylimion in Examples/viewORCA_multi/Example_2
username@computer:username/Desktop$ viewORCA multi Cu_II-BnzImine_1+.xyz --charge=+1
Number of electrons in Cu-BnzImine_1+.xyz is: 83
The total number of electrons in the system is odd.
The multiplicity can be an even number greater than or equal to 2 based on the M=2S+1 rule. Examples: 2,4,6,8,10,12,..


Example 3: Benzylamine
Benzylamine contains 58 electrons, so it can have an odd multiplicity. For organic systems, the ground spin state multiplicity is 1. See the xyz file for Benzylamine in Examples/viewORCA_multi/Example_3
username@computer:username/Desktop$ viewORCA multi Benzylamine.xyz --charge=0
Number of electrons in Benzylamine.xyz is: 58
The total number of electrons in the system is even.
The multiplicity can be an odd number greater than or equal to 1 based on the M=2S+1 rule. Examples: 1,3,5,7,9,11,..


Example 4: Cu(II)-Benzylimine
Cu(II)-Benzylimine contains 83 electrons, so it can have an even multiplicity. For Cu(II) systems, the ground spin state multiplicity is 2. See the xyz file for Cu(II)-Benzylimine in Examples/viewORCA_multi/Example_4
username@computer:username/Desktop$ viewORCA multi Cu_II-BnzImine-H_2+.xyz --charge=+2
Number of electrons in Cu-BnzImine-H_2+.xyz is: 83
The total number of electrons in the system is odd.
The multiplicity can be an even number greater than or equal to 2 based on the M=2S+1 rule. Examples: 2,4,6,8,10,12,..


Example 5: Benzylamion anion
Benzylamion anion (with a missing hydrogen and having a charge of -1) contains 58 electrons, so it can have an even multiplicity. For organic systems, the ground spin state multiplicity is 1. See the xyz file for Benzylamion anion in Examples/viewORCA_multi/Example_5
username@computer:username/Desktop$ viewORCA multi Benzyl-NH_1-.xyz --charge=-1
Number of electrons in Benzyl-NH_1-.xyz is: 58
The total number of electrons in the system is even.
The multiplicity can be an odd number greater than or equal to 1 based on the M=2S+1 rule. Examples: 1,3,5,7,9,11,..


viewORCA move_com: Move the Centre-of-Mass of your Chemical System¶
This method is designed to move the centre of mass of a chemical system.
To use this module:
- In the terminal, change directory into the directory containing the xyz files of the chemical systems you want to change the centre of masses of, and
-
type
viewORCA move_com filepath --move_centre_of_mass_to=move_centre_of_mass_tointo the terminal, where:filepathis the path to thexyzfile of your system, and--move_centre_of_mass_tois the position you would like to move the centre of mass of your chemical system to. Make sure you give your coordicates asx,y,z, including commas between coordinates. If--move_centre_of_mass_tois not given, the default given is the origin \((0.0,0.0,0.0)\).
The output file will be saved as filepath, but with move_centre_of_mass_to saved in the filename.
Example
Lets consider that you have a benzylamine molecule that you want to move its centre of mass to \((10.0,0.0,0.0)\). We perform the following in the terminal:
# First, change directory into the folder containing your xyz files you want to move the centre of mass of.
cd path_to_xyz_files
# Second, run "viewORCA move_com".
viewORCA move_com Benzylamine.xyz -c 10.0,0.0,0.0
We will get a new file called Benzylamine_com_10.0_0.0_0.0.xyz, which is the benzylamine molecule but moved to \((10.0,0.0,0.0)\). This example is given in Examples/viewORCA_move_com


viewORCA Combine: Combine Several Chemical Systems Together¶
This method is designed to allow the user to easily combine multiple xyz files together into the same chemical system.
To use this module:
- In the terminal, change directory into the directory containing the xyz files of the chemical systems you want to combine together.
-
type
viewORCA combine filepath_for_system_1 filepath_for_system_2 filepath_for_system_3 ...into the terminal, where:filepath_for_system_1, filepath_for_system_2, ...are the paths to all the xyz files you would like to combine together.
Example
Say we want to combine an xyz file containing a Cu(II)-benzylimine molecule (with centre of mass = \((0.0, 0.0, 0.0)\)) with another xyz file containing a benylamine molecule (with centre of mass = \((10.0, 0.0, 0.0)\)). We would run the code below to achieve this:
# First, change directory into the folder containing your xyz files you want to combine together.
cd path_to_xyz_files
# Second, run "viewORCA move_com".
viewORCA combine Benzylamine_com_10.0_0.0_0.0.xyz Cu_II-BnzImine_1+_com_0_0_0.xyz
viewORCA combine will print the shortest distances between the chemical system you combine together so you know they are a good distance from each other. If you see that any distance is less than 3.0 Å, you may have accidently merged your chemical systems together.
You will also see a new file has been made called overall_chemical_system.xyz that contains your overall chemical system. This example is given in Examples/viewORCA_combine. Make sure you check this file

